
Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues


 , and
, and
Abstract
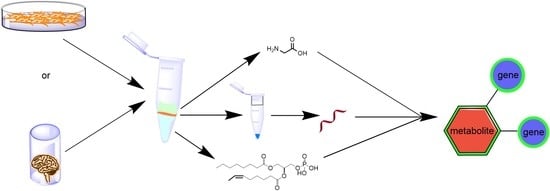
:
1. Introduction
2. Results and Discussion
2.1. Assessment of the Dual Extraction Methods for Metabolite/Lipid Extraction from Human Brain Cancer Cells
2.1.1. Metabolite Analysis
2.1.2. Lipids Analysis
2.2. Assessment of the Dual Extraction Methods for RNA Extraction from Human Brain Cancer Cells
2.3. The Effects of Acute Cellular Stress on Metabolism and Transcription in BXD-1425 Cells
2.4. Multi-Omics Integration
2.5. Application of Sequential/mirVana Method to Primary Brain Tumour Tissue
3. Materials and Methods
3.1. Preparation of Cell Pellets
3.2. Single Extraction of Metabolites or RNA
3.2.1. mirVana Kit
3.2.2. Sequential Solvent Addition and Shaking
3.3. Dual Extraction of Metabolites and RNA
3.3.1. Cryomill/mirVana: Cryomill and mirVana kit (Phenol-Chloroform and SPE)
3.3.2. Cryomill-wash/Econospin: Cryomill, Wash and Econospin Columns (SPE)
3.3.3. Rotation/Phenol-Chloroform: Rotate Cell Extracts and Phenol-Chloroform
3.3.4. Sequential/mirVana: Sequential Solvent Addition, Shake and mirVana Kit (Phenol-Chloroform and SPE)
3.4. Serum Starvation
3.5. LC-MS Sample Preparation
3.6. Applying Sequential/mirVana Method to Extract Metabolites, Lipids and RNA from Brain Tumour Tissue
3.7. LC-MS Analysis
3.8. Metabolite Identification
3.9. RNA and Transcriptomics Analysis
3.10. Statistical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kan, M.; Shumyatcher, M.; Himes, B.E. Using omics approaches to understand pulmonary diseases. Respir. Res. 2017, 18, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Glenn Tarcea, V.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futschik, M.E.; Morkel, M.; Schäfer, R.; Sers, C. The Human Transcriptome: Implications for Understanding, Diagnosing, and Treating Human Disease, 2nd ed.; Coleman, W.B., Tsongalis, G.J., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 135–164. [Google Scholar]
- Challacombe, J.F. In silico identification of metabolic enzyme drug targets in Burkholderia pseudomallei. bioRxiv 2017, 1–33. [Google Scholar] [CrossRef]
- Li, L.; Zhou, X.; Ching, W.K.; Wang, P. Predicting enzyme targets for cancer drugs by profiling human Metabolic reactions in NCI-60 cell lines. BMC Bioinform. 2010, 11, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, M.S.V.; Jackson, W.P.; Bailey, S.K.; Vayalil, P.; Landar, A.; Rostas, J.W.; Mulekar, M.S.; Samant, R.S.; Shevde, L.A. Deficiency of tumor suppressor Merlin facilitates metabolic adaptation by co-operative engagement of SMAD-Hippo signaling in breast cancer. Carcinogenesis 2018, 39, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Meierhofer, D.; Weidner, C.; Sauer, S. Integrative analysis of transcriptomics, proteomics, and metabolomics data of white adipose and liver tissue of high-fat diet and rosiglitazone-treated insulin-resistant mice identified pathway alterations and molecular hubs. J. Proteome Res. 2014, 13, 5592–5602. [Google Scholar] [CrossRef] [Green Version]
- Heiland, D.H.; Wörner, J.; Haaker, J.G.; Delev, D.; Pompe, N.; Mercas, B.; Franco, P.; Gäbelein, A.; Heynckes, S.; Pfeifer, D.; et al. The integrative metabolomic-transcriptomic landscape of glioblastome multiforme. Oncotarget 2017, 8, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Weckwerth, W.; Wenzel, K.; Fiehn, O. Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 2004, 4, 78–83. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS ONE 2015, 10, e0116740. [Google Scholar] [CrossRef]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of intra-Tumor Heterogeneity. Front. Med. 2018, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Leuthold, P.; Schwab, M.; Hofmann, U.; Winter, S.; Rausch, S.; Pollak, M.N.; Hennenlotter, J.; Bedke, J.; Schaeffeler, E.; Haag, M. Simultaneous Extraction of RNA and Metabolites from Single Kidney Tissue Specimens for Combined Transcriptomic and Metabolomic Profiling. J. Proteome Res. 2018, 17, 3039–3049. [Google Scholar] [CrossRef]
- Arzalluz-Luque, Á.; Devailly, G.; Mantsoki, A.; Joshi, A. Delineating biological and technical variance in single cell expression data. Int. J. Biochem. Cell Biol. 2017, 90, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Valledor, L.; Escandón, M.; Meijón, M.; Nukarinen, E.; Cañal, M.J.; Weckwerth, W. A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant. J. 2014, 79, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Vorreiter, F.; Richter, S.; Peter, M.; Baumann, S.; von Bergen, M.; Tomm, J.M. Comparison and optimization of methods for the simultaneous extraction of DNA, RNA, proteins, and metabolites. Anal. Biochem. 2016, 508, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roume, H.; Heintz-Buschart, A.; Muller, E.E.L.; Wilmes, P. Sequential Isolation of Metabolites, RNA, DNA, and Proteins from the Same Unique Sample, 1st ed.; DeLong, E.F., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 531, pp. 219–236. [Google Scholar]
- Hasegawa, Y.; Otoki, Y.; McClorry, S.; Coates, L.C.; Lombardi, R.L.; Taha, A.Y.; Slupsky, C.M. Optimization of a Method for the Simultaneous Extraction of Polar and Non-Polar Oxylipin Metabolites, DNA, RNA, Small RNA, and Protein from a Single Small Tissue Sample. Methods Protoc. 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- LoCoco, P.M.; Boyd, J.T.; Espitia Olaya, C.M.; Furr, A.R.; Garcia, D.K.; Weldon, K.S.; Zou, Y.; Locke, E.; Tobon, A.; Lai, Z.; et al. Reliable approaches to extract high-integrity RNA from skin and other pertinent tissues used in pain research. Pain Rep. 2020, 5, 818. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Muller, E.E.L.; Lebrun, L.A.; Wampach, L.; Wilmes, P. Sequential Isolation of DNA, RNA, Protein, and Metabolite Fractions from Murine Organs and Intestinal Contents for Integrated Omics of Host–Microbiota Interactions; Becher, D., Ed.; Springer: New York, NY, USA, 2018; Volume 1841, pp. 279–291. [Google Scholar]
- Ghawana, S.; Paul, A.; Kumar, H.; Kumar, A.; Singh, H.; Bhardwaj, P.K.; Rani, A.; Singh, R.S.; Raizada, J.; Singh, K.; et al. An RNA isolation system for plant tissues rich in secondary metabolites. BMC Res. Notes 2011, 4, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnaiyan, P.; Kensicki, E.; Bloom, G.; Prabhu, A.; Sarcar, B.; Kahali, S.; Eschrich, S.; Qu, X.; Forsyth, P.; Gillies, R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012, 72, 5878–5888. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.D.; Kohe, S.E.; Gill, S.K.; Davies, N.P.; Wilson, M.; Storer, L.C.D.; Ritzmann, T.; Paine, S.M.L.; Scott, I.S.; Nicklaus-Wollenteit, I.; et al. Tissue metabolite profiles for the characterisation of paediatric cerebellar tumours. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Cuellar-Baena, S.; Morales, J.M.; Martinetto, H.; Calvar, J.; Sevlever, G.; Castellano, G.; Cerda-Nicolas, M.; Celda, B.; Monleon, D. Comparative metabolic profiling of paediatric ependymoma, medulloblastoma and pilocytic astrocytoma. Int. J. Mol. Med. 2010, 26, 941–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Promyslov, M.S.; Mirzoian, R.A. Homocarnosine metabolism in brain tissue. Vopr. Biokhimii Mozga 1976, 11, 71–77. [Google Scholar]
- Woolman, M.; Ferry, I.; Kuzan-Fischer, C.M.; Wu, M.; Zou, J.; Kiyota, T.; Isik, S.; Dara, D.; Aman, A.; Das, S.; et al. Rapid determination of medulloblastoma subgroup affiliation with mass spectrometry using a handheld picosecond infrared laser desorption probe. Chem. Sci. 2017, 8, 6508–6519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Villanueva, N.; Bittar, T.; Arsenault, E.; Labonté, B.; Huan, T. Parallel metabolomics and lipidomics enables the comprehensive study of mouse brain regional metabolite and lipid patterns. Anal. Chim. Acta 2020, 1136, 168–177. [Google Scholar] [CrossRef]
- Wood, J.A. Identifying the Metabolic ‘Achilles Heel’ of Adult and Paediatric Glioblastoma Multiforme; The University of Nottingham: Nottingham, UK, 2018. [Google Scholar]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Hutschenreuther, A.; Kiontke, A.; Birkenmeier, G.; Birkemeyer, C. Comparison of extraction conditions and normalization approaches for cellular metabolomics of adherent growing cells with GC-MS. Anal. Methods 2012, 4, 1953–1963. [Google Scholar] [CrossRef]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, D.Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef]
- Wu, H.; Southam, A.D.; Hines, A.; Viant, M.R. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal. Biochem. 2008, 372, 204–212. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metab. 2013, 1, 92–107. [Google Scholar] [CrossRef]
- Wheelock, Å.M.; Wheelock, C.E. Trials and tribulations of ‘omics data analysis: Assessing quality of SIMCA-based multivariate models using examples from pulmonary medicine. Mol. Biosyst. 2013, 9, 2589. [Google Scholar] [CrossRef] [Green Version]
- Halama, A.; Riesen, N.; Möller, G.; Hrabě de Angelis, M.; Adamski, J. Identification of biomarkers for apoptosis in cancer cell lines using metabolomics: Tools for individualized medicine. J. Intern. Med. 2013, 274, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ye, H.; Shao, C.; Zheng, X.; Li, Q.; Wang, L.; Zhao, M.; Lu, G.; Chen, B.; Zhang, J.; et al. Metabolomics–Proteomics Combined Approach Identifies Differential Metabolism-Associated Molecular Events between Senescence and Apoptosis. J. Proteome Res. 2017, 16, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Wang, K.; He, J.; Qiu, Y.; Xie, G.; Su, M.; Jia, W.; Li, H. Effects of ADMA on gene expression and metabolism in serum-starved LoVo cells. Sci. Rep. 2016, 6, 25892. [Google Scholar] [CrossRef] [Green Version]
- Nestel, P.J.; Couzens, E.A. Turnover of Individual Cholesterol Esters in Human Liver and Plasma. J. Clin. Investig. 1966, 45, 1234–1240. [Google Scholar] [CrossRef]
- Jumpsen, J.A.; Lien, E.L.; Goh, Y.K.; Clandinin, M.T. During neuronal and glial cell development diet n−6 to n−3 fatty acid ratio alters the fatty acid composition of phosphatidylinositol and phosphatidylserine. Biochim. Biophys. Acta Lipids Lipid Metab. 1997, 1347, 40–50. [Google Scholar] [CrossRef]
- Morales, A.; Lee, H.; Goñi, F.M.; Kolesnick, R.; Fernandez-Checa, J.C. Sphingolipids and cell death. Apoptosis 2007, 12, 923–939. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 7th ed.; Macmillan Higher Education: Basingstoke, UK, 2017. [Google Scholar]
- Sun, J. Metabolomics in Drug-induced Toxicity and Drug Metabolism. J. Drug Metab. Toxicol. 2012, 3, 1–2. [Google Scholar] [CrossRef]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Chibalin, A.V. Serum starvation: Caveat emptor. Am. J. Physiol. Cell Physiol. 2011, 301, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Pecqueur, C.; Oliver, L.; Oizel, K.; Lalier, L.; Vallette, F.M. Targeting Metabolism to Induce Cell Death in Cancer Cells and Cancer Stem Cells. Int. J. Cell Biol. 2013, 2013, 805975. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Baxter, P.A.; Voicu, H.; Gurusiddappa, S.; Zhao, Y.; Adesina, A.; Man, T.K.; Shu, Q.; Zhang, Y.J.; Zhao, X.M.; et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro-Oncology 2010, 12, 580–594. [Google Scholar] [CrossRef]
- Jung, Y.D.; Nakano, K.; Liu, W.; Gallick, G.E.; Ellis, L.M. Extracellular signal-regulated kinase activation is required for up-regulation of vascular endothelial growth factor by serum starvation in human colon carcinoma cells. Cancer Res. 1999, 59, 4804–4807. [Google Scholar]
- Schatschneider, S.; Abdelrazig, S.; Safo, L.; Henstra, A.M.; Millat, T.; Kim, D.-H.; Winzer, K.; Minton, N.P.; Barrett, D.A. Quantitative Isotope-Dilution High-Resolution-Mass-Spectrometry Analysis of Multiple Intracellular Metabolites in Clostridium autoethanogenum with Uniformly 13C-Labeled Standards Derived from Spirulina. Anal. Chem. 2018, 90, 4470–4477. [Google Scholar] [CrossRef]
- Abuawad, A.; Mbadugha, C.; Ghaemmaghami, A.M.; Kim, D.H. Metabolic characterisation of THP-1 macrophage polarisation using LC–MS-based metabolite profiling. Metabolomics 2020, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Pousinis, P.; Gowler, P.R.W.; Burston, J.J.; Ortori, C.A.; Chapman, V.; Barrett, D.A. Lipidomic identification of plasma lipids associated with pain behaviour and pathology in a mouse model of osteoarthritis. Metabolomics 2020, 16, 32. [Google Scholar] [CrossRef] [Green Version]
- Tautenhahn, R.; Bottcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Scheltema, R.A.; Jankevics, A.; Jansen, R.C.; Swertz, M.A.; Breitling, R. PeakML/mzMatch: A file format, Java library, R library, and tool-chain for mass spectrometry data analysis. Anal. Chem. 2011, 83, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E.V. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography-mass spectrometry: Improved metabolite identification by retention time prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Jankevics, A.; Burgess, K.E.V.; Breitling, R.; Barrett, M.P. IDEOM: An Excel interface for analysis of LC–MS-based metabolomics data. Bioinformatics 2012, 28, 1048–1049. [Google Scholar] [CrossRef]
- Sumner, L.W.; Lei, Z.; Nikolau, B.J.; Saito, K.; Roessner, U.; Trengove, R. Proposed quantitative and alphanumeric metabolite identification metrics. Metabolomics 2014, 10, 1047–1049. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- William, S. The probable error of a mean. Biometrika 1908, 6, 1–25. [Google Scholar] [CrossRef]
- Fisher, R.A. The Correlation Between Relatives on the Supposition of Mendelian Inheritance. Philos. Trans. Royal Soc. Edinb. 1918, 52, 399–433. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extraction Method | Sequential Solvent Addition and Shaking (Positive Control) | Cryomill/mirVana | Cryomill-Wash/Econospin | Rotation/Phenol-chloroform | Sequential/mirVana |
|---|---|---|---|---|---|
| n | 3 | 2 | 4 | 3 | 4 |
| Total ion count | 4.95 × 107 ± 1.53 × 106 | 5.58 × 107 ± 2.17 × 106 | 5.80 × 107 ± 3.66 × 106 * | 5.35 × 107 ± 8.97 × 106 | 5.01 × 107 ± 5.50 × 106 |
| Number of ions (unfiltered) | 2025 ± 2 | 2022 ± 1 | 2024 ± 2 | 2013 ± 5 | 2025 ± 3 |
| Number of metabolites | 193 | 193 | 193 | 193 | 193 |
| Significantly altered metabolites * | - | 31 (22 ↑, 9 ↓) | 0 | 1 ↓ | 0 |
| Practical comments | -Cryo-milling can damage tubes and leak solvents –Quick protocol which can use any RNA extraction kit. | -Cryo-milling can damage tubes and leak solvents. -Long protocol involving washing steps purifies metabolites but is time-consuming. Cheap, but time-consuming RNA extraction as uses self-made buffers. | Metabolite extraction protocol is simple but is not biphasic so cannot perform lipidomics on a C18 column. | Easy to perform, possibility to extract microRNA separately. Longest solvent evaporation time. |
| Extraction Method | Sequential Solvent Addition and Shaking (Positive Control) | Cryomill/mirVana | Cryomill-Wash/Econospin | Sequential/mirVana |
|---|---|---|---|---|
| n | 3 | 2 | 4 | 4 |
| Total ion count—negative | 3.41 × 109 ± 1.44 × 108 | 3.36 × 109 ± 1.80 × 108 | 3.74 × 109 ± 3.35 × 108 | 3.45 × 109 ± 3.52 × 108 |
| Total ion count—positive | 7.12 × 109 ± 2.33 × 108 | 6.48 × 109 ± 4.33 × 108 | 7.03 × 109 ± 6.04 × 108 | 7.40 × 109 ± 7.28 × 108 |
| Number of putative lipids | 3390 | 3428 | 3506 | 3427 |
| Significantly altered lipids * | - | 356 (256 ↑, 100 ↓) | 618 (394 ↑, 224 ↓) | 314 (244 ↑, 70 ↓) |
| Extraction Method | mirVana Kit (Positive Control) | Cryomill/mirVana | Cryomill-Wash/Econospin | Rotation/Phenol-Chloroform | Sequential/mirVana |
|---|---|---|---|---|---|
| Concentration of RNA (ng/µL) | 1300 ± 21 | 910 ± 26 * | 514 ± 29 * | 1331 ± 249 | 887 ± 131 * |
| Mass of RNA (µg) | 65.0 ± 1.3 | 45.5 ± 1.8 * | 25.7 ± 2.0 * | 20.0 ± 4.6 * | 44.4 ± 7.6 * |
| Relative % of RNA recovered | 100 | 70* | 40 * | 31 * | 68 * |
| 260/280 ratio | 2.10 ±0.01 | 2.05 ±0.01 | 2.16 ±0.01 * | 2.11 ±0.01 | 2.06 ±0.01 * |
| 260/230 ratio | 1.80±0.47 a | 2.03 ±0.15 | 1.97 ±0.02 | 2.09 ±0.02 | 2.05 ±0.07 |
| 28S/18S (Area) | 2.9 ± 0.1 | 2.6 ± 0.1 | 2.7 ± 0.1 | 1.4 ± 0.1 * | 2.2 ± 0.7 |
| RIN | 9.4 ± 0.1 | 7.9 ± 0.5 | 9.1 ± 0.2 | 7.6 ± 0.3 * | 8.9 ± 0.1 * |
| Gene | Metabolic Pathway | p-Value | Fold-Change |
|---|---|---|---|
| ATP1A1 | Purine metabolism | 1.69 × 105 | 1.37693 |
| AURKA | Glycosphingolipid metabolism | 6.37 × 105 | −4.80815 |
| BUB1 | Glycosphingolipid metabolism | 1.39 × 104 | −4.57951 |
| BUB1B | Glycosphingolipid metabolism | 1.64 × 105 | −3.05730 |
| CDC25C | Glycosphingolipid metabolism | 6.00 × 105 | −2.45823 |
| CTPS1 | Pyrimidine metabolism | 2.03 × 104 | −2.73359 |
| CYP1B1 | Arachidonic acid metabolism | 2.18 × 105 | 4.91775 |
| DDAH1 | Arginine, proline, glutamate, aspartate and asparagine metabolism | 4.29 × 105 | −2.61779 |
| DHFR | Folate metabolism | 1.76 × 104 | −1.89205 |
| DUSP6 | Glycosphingolipid metabolism | 1.98 × 104 | 3.58612 |
| FADS3 | Omega-6 fatty acid metabolism | 2.04 × 104 | −2.85552 |
| FMO3 | Amino sugars metabolism | 2.41 × 104 | 1.80707 |
| GUCY1B3 | Purine metabolism | 1.36 × 105 | 10.77960 |
| NDST3 | Proteoglycan metabolism | 6.77 × 105 | 1.60250 |
| NDUFB2 | Methionine metabolism | 1.27 × 104 | −1.47355 |
| P4HA1 | Arginine, proline, glutamate, aspartate and asparagine metabolism | 1.73 × 104 | 3.91765 |
| PLPP3 | Glycerophospholipid metabolism | 7.39 × 106 | 2.52901 |
| PTGDS | Prostaglandin formation from arachidonate | 1.55 × 104 | 2.43688 |
| RFC5 | Purine metabolism | 2.00 × 104 | −2.96426 |
| SAT1 | Arginine, proline, glutamate, aspartate and asparagine metabolism | 6.22 × 106 | 6.06009 |
| TGFBR2 | Glycosphingolipid metabolism | 1.11 × 104 | 2.51642 |
| TRMU | Methionine metabolism | 2.30 × 104 | −2.36320 |
| TYMS | Folate metabolism | 1.71 × 104 | −3.64677 |
| UBE2C | Purine metabolism | 2.10 × 104 | −6.48322 |
| UBE2T | Purine metabolism | 3.33 × 106 | −3.90861 |
| WHSC1 | Lysine metabolism | 7.95 × 105 | −2.65510 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodward, A.; Pandele, A.; Abdelrazig, S.; Ortori, C.A.; Khan, I.; Uribe, M.C.; May, S.; Barrett, D.A.; Grundy, R.G.; Kim, D.-H.; et al. Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues. Metabolites 2021, 11, 240. https://doi.org/10.3390/metabo11040240
Woodward A, Pandele A, Abdelrazig S, Ortori CA, Khan I, Uribe MC, May S, Barrett DA, Grundy RG, Kim D-H, et al. Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues. Metabolites. 2021; 11(4):240. https://doi.org/10.3390/metabo11040240
Chicago/Turabian StyleWoodward, Alison, Alina Pandele, Salah Abdelrazig, Catherine A. Ortori, Iqbal Khan, Marcos Castellanos Uribe, Sean May, David A. Barrett, Richard G. Grundy, Dong-Hyun Kim, and et al. 2021. "Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues" Metabolites 11, no. 4: 240. https://doi.org/10.3390/metabo11040240
APA StyleWoodward, A., Pandele, A., Abdelrazig, S., Ortori, C. A., Khan, I., Uribe, M. C., May, S., Barrett, D. A., Grundy, R. G., Kim, D. -H., & Rahman, R. (2021). Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues. Metabolites, 11(4), 240. https://doi.org/10.3390/metabo11040240