
Multi-Omics-Based Discovery of Plant Signaling Molecules


{kind=link}
{kind=link}
Abstract
:1. Introduction
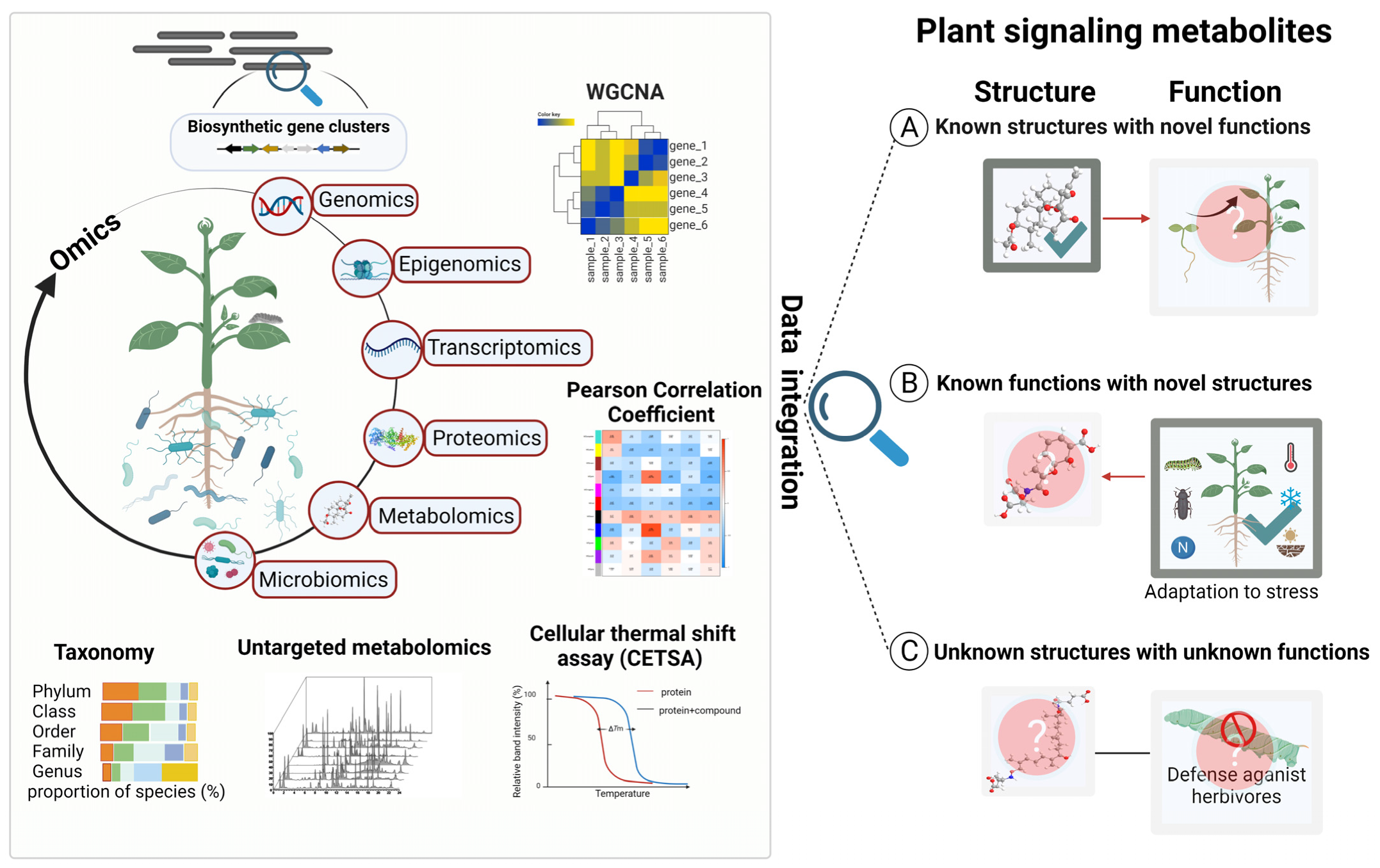
2. Multi-Omics as a Powerful Tool for Uncovering Plant Signaling Metabolites
2.1. Features of Multi-Omics, Including Genomics, Epigenomics, Transcriptomics, Proteomics, Metabolomics and Microbiomics
2.1.1. Genomics—The Source Code for Discovering Plant Signaling Metabolites
2.1.2. Epigenomics—The Gatekeeper for Plant Metabolite Biosynthesis
2.1.3. Transcriptomics—Snapshots of Gene Expression under Specific Spatial–Temporal Conditions
2.1.4. Proteomics—The Yet to Flourish Tool for Plant Signaling Metabolite Discovery
2.1.5. Metabolomics—The Node of Multi-Omics for Discovering Signaling Metabolites
2.1.6. Microbiomics—Uncovering Metabolite and Microbe Interactions
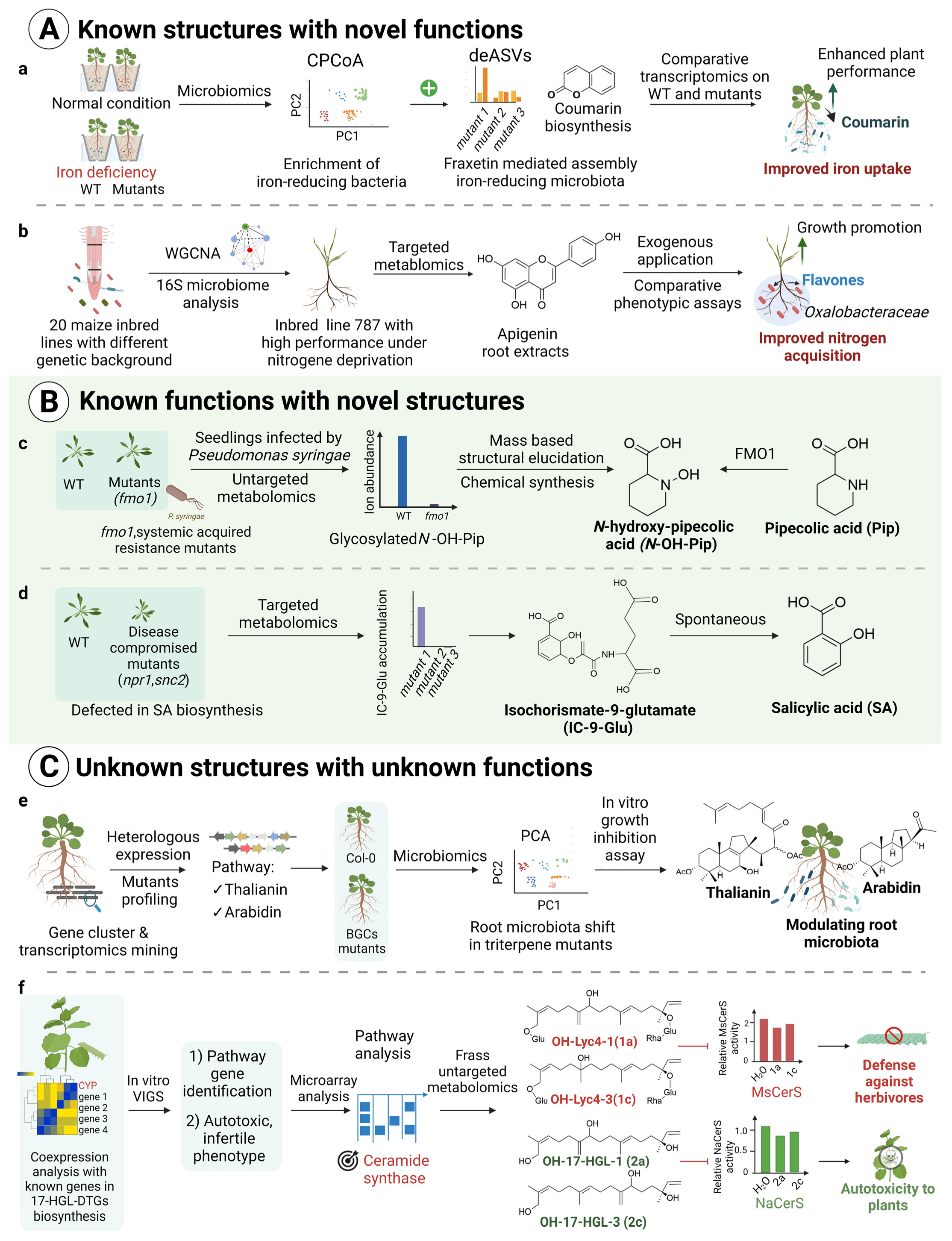
2.2. Multi-Omics-Based Discovery of New Functions of Known Molecules
2.3. Multi-Omics-Based Discovery of Unknown Molecules with Known Functions
2.4. Multi-Omics-Based Discovery of Unknown Molecules with Unknown Functions
3. Breaking the Limitation of Multi-Omics: Future Perspective for Accelerated Discovery of Plant Signaling Molecules
Author Contributions
Funding
Conflicts of Interest
References
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. Knapsack family databases: Integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [Green Version]
- Kessler, A.; Kalske, A. Plant secondary metabolite diversity and species interactions. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 115–138. [Google Scholar] [CrossRef]
- Ballare, C.L. Jasmonate-induced defenses: A tale of intelligence, collaborators and rascals. Trends Plant Sci. 2011, 16, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, K.; Upadhyay, N.; Kumar, N.; Yadav, G.; Singh, J.; Mishra, R.K.; Kumar, V.; Verma, R.; Upadhyay, R.G.; Pandey, M.; et al. Abscisic acid signaling and abiotic stress tolerance in plants: A review on current knowledge and future prospects. Front. Plant Sci. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Planas-Riverola, A.; Gupta, A.; Betegón-Putze, I.; Bosch, N.; Ibañes, M.; Caño-Delgado, A.I. Brassinosteroid signaling in plant development and adaptation to stress. Development 2019, 146, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-Y.; Sae-Seaw, J.; Wang, Z.-Y. Brassinosteroid signalling. Development 2013, 140, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Teale, W.D.; Paponov, I.A.; Palme, K. Auxin in action: Signalling, transport and the control of plant growth and development. Nat. Rev. Mol. Cell Biol. 2006, 7, 847–859. [Google Scholar] [CrossRef]
- Sun, T.P.; Gubler, F. Molecular mechanism of gibberellin signaling in plants. Annu. Rev. Plant Biol. 2004, 55, 197–223. [Google Scholar] [CrossRef] [Green Version]
- Brewer, P.B.; Koltai, H.; Beveridge, C.A. Diverse roles of strigolactones in plant development. Mol. Plant 2013, 6, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Binder, B.M. Ethylene signaling in plants. J. Biol. Chem. 2020, 295, 7710–7725. [Google Scholar] [CrossRef] [Green Version]
- Rivas-San Vicente, M.; Plasencia, J. Salicylic acid beyond defence: Its role in plant growth and development. J. Exp. Bot. 2011, 62, 3321–3338. [Google Scholar] [CrossRef] [Green Version]
- Kemen, A.C.; Honkanen, S.; Melton, R.E.; Findlay, K.C.; Mugford, S.T.; Hayashi, K.; Haralampidis, K.; Rosser, S.J.; Osbourn, A. Investigation of triterpene synthesis and regulation in oats reveals a role for β-amyrin in determining root epidermal cell patterning. Proc. Natl. Acad. Sci. USA 2014, 111, 8679–8684. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Fernandez-Calvo, P.; Ritter, A.; Huang, A.C.; Morales-Herrera, S.; Bicalho, K.U.; Karady, M.; Pauwels, L.; Buyst, D.; Njo, M.; et al. Modulation of Arabidopsis root growth by specialized triterpenes. New Phytol. 2021, 230, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Nisani, S.; Yadav, B.S.; Woldemariam, M.G.; Shai, B.; Obolski, U.; Ehrlich, M.; Shani, E.; Jander, G.; Chamovitz, D.A. The glucosinolate breakdown product indole-3-carbinol acts as an auxin antagonist in roots of Arabidopsis thaliana. Plant J. 2015, 82, 547–555. [Google Scholar] [CrossRef]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; van der Heijden, M.G.A.; et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 2738–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nützmann, H.-W.; Huang, A.; Osbourn, A. Plant metabolic clusters—From genetics to genomics. New Phytol. 2016, 211, 771–789. [Google Scholar] [CrossRef] [Green Version]
- Meena, K.K.; Sorty, A.M.; Bitla, U.M.; Choudhary, K.; Gupta, P.; Pareek, A.; Singh, D.P.; Prabha, R.; Sahu, P.K.; Gupta, V.K.; et al. Abiotic stress responses and microbe-mediated mitigation in plants: The omics strategies. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Chu, L.; Huang, J.; Muhammad, M.; Deng, Z.; Gao, J. Genome mining as a biotechnological tool for the discovery of novel marine natural products. Crit. Rev. Biotechnol. 2020, 40, 571–589. [Google Scholar] [CrossRef]
- Kersey, P.J. Plant genome sequences: Past, present, future. Curr. Opin. Plant Biol. 2019, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.G.; Suarez, D.H.; Kai, B.; Anne, O.; Medema, M.H. Plantismash: Automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, W55–W63. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Kautsar, S.A.; Hong, Y.J.; Medema, M.H.; Bond, A.D.; Tantillo, D.J.; Osbourn, A. Unearthing a sesterterpene biosynthetic repertoire in the Brassicaceae through genome mining reveals convergent evolution. Proc. Natl. Acad. Sci. USA 2017, 114, E6005–E6014. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Hong, Y.J.; Bond, A.D.; Tantillo, D.J.; Osbourn, A. Diverged plant terpene synthases reroute the carbocation cyclization path towards the formation of unprecedented 6/11/5 and 6/6/7/5 sesterterpene scaffolds. Angew. Chem. Int. Ed. 2018, 57, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Kersten, R.D.; Weng, J.-K. Gene-guided discovery and engineering of branched cyclic peptides in plants. Proc. Natl. Acad. Sci. USA 2018, 115, E10961–E10969. [Google Scholar] [CrossRef] [Green Version]
- Töpfer, N.; Fuchs, L.-M.; Aharoni, A. The phytoclust tool for metabolic gene clusters discovery in plant genomes. Nucleic Acids Res. 2017, 45, 7049–7063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schläpfer, P.; Zhang, P.; Wang, C.; Kim, T.; Banf, M.; Chae, L.; Dreher, K.; Chavali, A.K.; Nilo-Poyanco, R.; Bernard, T.; et al. Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 2017, 173, 2041–2059. [Google Scholar] [CrossRef] [Green Version]
- Chavali, A.K.; Rhee, S.Y. Bioinformatics tools for the identification of gene clusters that biosynthesize specialized metabolites. Brief. Bioinform. 2018, 19, 1022–1034. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Nuetzmann, H.-W.; MacDonald, J.T.; Moore, B.; Field, B.; Berriri, S.; Trick, M.; Rosser, S.J.; Kumar, S.V.; Freemont, P.S.; et al. Delineation of metabolic gene clusters in plant genomes by chromatin signatures. Nucleic Acids Res. 2016, 44, 2255–2265. [Google Scholar] [CrossRef]
- Zhao, K.; Kong, D.; Jin, B.; Smolke, C.D.; Rhee, S.Y. A novel bivalent chromatin associates with rapid induction of camalexin biosynthesis genes in response to a pathogen signal in Arabidopsis. eLife 2021, 10, e69508. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Lei, L.; Liu, Z.; Zhou, S.; Yang, C.; Zhu, X.; Guo, H.; Zhang, F.; Peng, M.; Zhang, M.; et al. Selection of a subspecies-specific diterpene gene cluster implicated in rice disease resistance. Nat. Plants 2020, 6, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- van Gurp, T.P.; Wagemaker, N.C.A.M.; Wouters, B.; Vergeer, P.; Ouborg, J.N.J.; Verhoeven, K.J.F. epiGBS: Reference-free reduced representation bisulfite sequencing. Nat. Methods 2016, 13, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nett, R.S.; Lau, W.; Sattely, E.S. Discovery and engineering of colchicine alkaloid biosynthesis. Nature 2020, 584, 148–153. [Google Scholar] [CrossRef]
- Hodgson, H.; De La Peña, R.; Stephenson, M.J.; Thimmappa, R.; Vincent, J.L.; Sattely, E.S.; Osbourn, A. Identification of key enzymes responsible for protolimonoid biosynthesis in plants: Opening the door to azadirachtin production. Proc. Natl. Acad. Sci. USA 2019, 116, 17096–17104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, L.; Kim, T.; Nilo-Poyanco, R.; Rhee, S.Y. Genomic signatures of specialized metabolism in plants. Science 2014, 344, 510–513. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, F. Hierarchical clustering. In Introduction to HPC with MPI for Data Science; Springer: Cham, Switzerland, 2016; pp. 195–211. [Google Scholar]
- Usadel, B.O.T.; Mutwil, M.; Giorgi, F.M.; Bassel, G.W.; Tanimoto, M.; Chow, A.; Steinhauser, D.; Persson, S.; Provart, N.J. Co-expression tools for plant biology: Opportunities for hypothesis generation and caveats. Plant Cell Environ. 2009, 32, 1633–1651. [Google Scholar] [CrossRef]
- Julca, I.; Ferrari, C.; Flores-Tornero, M.; Proost, S.; Lindner, A.-C.; Hackenberg, D.; Steinbachová, L.; Michaelidis, C.; Gomes Pereira, S.; Misra, C.S.; et al. Comparative transcriptomic analysis reveals conserved programmes underpinning organogenesis and reproduction in land plants. Nat. Plants 2021, 7, 1143–1159. [Google Scholar] [CrossRef]
- Obayashi, T.; Kinoshita, K. Rank of correlation coefficient as a comparable measure for biological significance of gene coexpression. DNA Res. 2009, 16, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Wehrens, R.; Buydens, L.M.C. Self- and super-organizing maps in R: The kohonen package. J. Stat. Softw. 2007, 21, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wisecaver, J.H.; Borowsky, A.T.; Tzin, V.; Jander, G.; Kliebenstein, D.J.; Rokas, A. A global coexpression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 2017, 29, 944–959. [Google Scholar] [CrossRef] [Green Version]
- Orme, A.; Louveau, T.; Stephenson, M.J.; Appelhagen, I.; Melton, R.; Cheema, J.; Li, Y.; Zhao, Q.; Zhang, L.; Fan, D.; et al. A noncanonical vacuolar sugar transferase required for biosynthesis of antimicrobial defense compounds in oat. Proc. Natl. Acad. Sci. USA 2019, 116, 27105–27114. [Google Scholar] [CrossRef] [Green Version]
- Sulis, D.B.; Wang, J.P. Regulation of lignin biosynthesis by post-translational protein modifications. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef]
- Kourelis, J.; Kaschani, F.; Grosse-Holz, F.M.; Homma, F.; Kaiser, M.; van der Hoorn, R.A.L. A homology-guided, genome-based proteome for improved proteomics in the alloploid Nicotiana benthamiana. BMC Genom. 2019, 20, 722. [Google Scholar] [CrossRef]
- Prabhu, N.; Dai, L.; Nordlund, P. CETSA in integrated proteomics studies of cellular processes. Curr. Opin. Chem. Biol. 2020, 54, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Venegas-Molina, J.; Molina-Hidalgo, F.J.; Clicque, E.; Goossens, A. Why and how to dig into plant metabolite-protein interactions. Trends Plant Sci. 2021, 26, 472–483. [Google Scholar] [CrossRef]
- Murale, D.P.; Hong, S.C.; Haque, M.M.; Lee, J.-S. Photo-affinity labeling (PAL) in chemical proteomics: A handy tool to investigate protein-protein interactions (PPIS). Proteome Sci. 2017, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Esteso, M.J.; Martínez-Márquez, A.; Sellés-Marchart, S.; Morante-Carriel, J.A.; Bru-Martínez, R. The role of proteomics in progressing insights into plant secondary metabolism. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [Green Version]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mergner, J.; Frejno, M.; List, M.; Papacek, M.; Chen, X.; Chaudhary, A.; Samaras, P.; Richter, S.; Shikata, H.; Messerer, M.; et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 2020, 579, 409–414. [Google Scholar] [CrossRef]
- Decker, G.; Wanner, G.; Zenk, M.H.; Lottspeich, F. Characterization of proteins in latex of the opium poppy (Papaver somniferum) using two-dimensional gel electrophoresis and microsequencing. Electrophoresis 2000, 21, 3500–3516. [Google Scholar] [CrossRef]
- Batista, A.N.L.; Santos-Pinto, J.R.A.d.; Batista, J.M.; Souza-Moreira, T.M.; Santoni, M.M.; Zanelli, C.F.; Kato, M.J.; López, S.N.; Palma, M.S.; Furlan, M. The combined use of proteomics and transcriptomics reveals a complex secondary metabolite network in Peperomia obtusifolia. J. Nat. Prod. 2017, 80, 1275–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Su, C.; Du, X.; Wang, R.; Chen, S.; Zhou, Y.; Liu, C.; Liu, X.; Tian, R.; Zhang, L.; et al. FAD-dependent enzyme-catalysed intermolecular 4+2 cycloaddition in natural product biosynthesis. Nat. Chem. 2020, 12, 620–628. [Google Scholar] [CrossRef]
- Redding-Johanson, A.M.; Batth, T.S.; Chan, R.; Krupa, R.; Szmidt, H.L.; Adams, P.D.; Keasling, J.D.; Lee, T.S.; Mukhopadhyay, A.; Petzold, C.J. Targeted proteomics for metabolic pathway optimization: Application to terpene production. Metab. Eng. 2011, 13, 194–203. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Beale, D.J.; Pinu, F.R.; Kouremenos, K.A.; Poojary, M.M.; Narayana, V.K.; Boughton, B.A.; Kanojia, K.; Dayalan, S.; Jones, O.A.H.; Dias, D.A. Review of recent developments in GC–MS approaches to metabolomics-based research. Metabolomics 2018, 14, 152. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Stassen, M.J.J.; Hsu, S.-H.; Pieterse, C.M.J.; Stringlis, I.A. Coumarin communication along the microbiome–root–shoot axis. Trends Plant Sci. 2021, 26, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Wurtzel, E.T.; Kutchan, T.M. Plant metabolism, the diverse chemistry set of the future. Science 2016, 353, 1232–1236. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-K. Translational genomics and multi-omics integrated approaches as a useful strategy for crop breeding. Genes Genom. 2019, 41, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Ke, L.; Zhang, D.; Wu, Y.; Sun, Y.; Mei, J.; Sun, J.; Sun, Y. Multi-omics assisted identification of the key and species-specific regulatory components of drought-tolerant mechanisms in Gossypium stocksii. Plant Biotechnol. J. 2021, 19, 1690–1692. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Jing, Y.; Lin, S.; Yue, Z.; Yang, X.; Xu, J.; Wu, J.; Zhang, Z.; Xia, R.; Zhu, J.; et al. Polyploidy underlies co-option and diversification of biosynthetic triterpene pathways in the apple tribe. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Qing, Z.; Liu, J.; Yi, X.; Liu, X.; Hu, G.; Lao, J.; He, W.; Yang, Z.; Zou, X.; Sun, M.; et al. The chromosome-level Hemerocallis citrina borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic. Res.-Engl. 2021, 8, 89. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, X.; Liu, X.; Zhu, X.; Li, Z.; Chu, H.; Wang, Q.; Lou, Q.; Cai, B.; Yang, Y.; et al. Chromosome-level genome of Himalayan yew provides insights into the origin and evolution of the paclitaxel biosynthetic pathway. Mol. Plant 2021, 14, 1199–1209. [Google Scholar] [CrossRef]
- Hou, S.; Wolinska, K.W.; Hacquard, S. Microbiota-root-shoot-environment axis and stress tolerance in plants. Curr. Opin. Plant Biol. 2021, 62, 102028. [Google Scholar] [CrossRef]
- Hou, S.; Thiergart, T.; Vannier, N.; Mesny, F.; Ziegler, J.; Pickel, B.; Hacquard, S. A microbiota-root-shoot circuit favours Arabidopsis growth over defence under suboptimal light. Nat. Plants 2021, 7, 1078–1092. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Chiniquy, D.; Pierroz, G.; Deng, S.; Gao, C.; Diamond, S.; Simmons, T.; Wipf, H.M.L.; Caddell, D.; et al. Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics. Nat. Commun. 2021, 12, 3209–3226. [Google Scholar] [CrossRef] [PubMed]
- Claassens, A.P.; Hills, P.N. Effects of strigolactones on plant roots. In Root Biology. Soil Biology; Springer: Cham, Switzerland, 2018; pp. 43–63. [Google Scholar]
- Cadot, S.; Guan, H.; Bigalke, M.; Walser, J.-C.; Jander, G.; Erb, M.; van der Heijden, M.G.A.; Schlaeppi, K. Specific and conserved patterns of microbiota-structuring by maize benzoxazinoids in the field. Microbiome 2021, 9, 103–122. [Google Scholar] [CrossRef]
- Harbort, C.J.; Hashimoto, M.; Inoue, H.; Niu, Y.; Guan, R.; Rombola, A.D.; Kopriva, S.; Voges, M.; Sattely, E.S.; Garrido-Oter, R.; et al. Root-secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis. Cell Host Microbe 2020, 28, 825–837.e826. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Holmes, E.C.; Rajniak, J.; Kim, J.-G.; Tang, S.; Fischer, C.R.; Mudgett, M.B.; Sattely, E.S. N-hydroxy-pipecolic acid is a mobile metabolite that induces systemic disease resistance in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E4920–E4929. [Google Scholar] [CrossRef] [Green Version]
- Rekhter, D.; Lüedke, D.; Ding, Y.; Feussner, K.; Zienkiewicz, K.; Lipka, V.; Wiermer, M.; Zhang, Y.; Feussner, I. Isochorismate-derived biosynthesis of the plant stress hormone salicylic acid. Science 2019, 365, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Jiang, T.; Liu, Y.X.; Bai, Y.C.; Reed, J.; Qu, B.; Goossens, A.; Nutzmann, H.W.; Bai, Y.; Osbourn, A. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 2019, 364, 546–556. [Google Scholar] [CrossRef]
- Li, J.; Halitschke, R.; Li, D.; Paetz, C.; Su, H.; Heiling, S.; Xu, S.; Baldwin, I.T. Controlled hydroxylations of diterpenoids allow for plant chemical defense without autotoxicity. Science 2021, 371, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Stringlis, I.A.; de Jonge, R.; Pieterse, C.M.J. The age of coumarins in plant-microbe interactions. Plant Cell Physiol. 2019, 60, 1405–1419. [Google Scholar] [CrossRef] [Green Version]
- Stringlis, I.A.; Yu, K.; Feussner, K.; de Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.; Feussner, I.; Pieterse, C.M.J. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudjordjie, E.N.; Sapkota, R.; Steffensen, S.K.; Fomsgaard, I.S.; Nicolaisen, M. Maize synthesized benzoxazinoids affect the host associated microbiome. Microbiome 2019, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Doseff, A.I.; Grotewold, E. Flavones: From biosynthesis to health benefits. Plants 2016, 5, 27. [Google Scholar] [CrossRef]
- Rajniak, J.; Barco, B.; Clay, N.K.; Sattely, E.S. A new cyanogenic metabolite in Arabidopsis required for inducible pathogen defence. Nature 2015, 525, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.E.; Kim, J.G.; Fischer, C.R.; Mehta, N.; Dufour-Schroif, C.; Wemmer, K.; Mudgett, M.B.; Sattely, E. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell 2020, 180, 176–187.e119. [Google Scholar] [CrossRef]
- Mishina, T.E.; Zeier, J.R. The Arabidopsis flavin-dependent monooxygenase fmo1 is an essential component of biologically induced systemic acquired resistance. Plant Physiol. 2006, 141, 1666–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.C.; Chen, Y.-C.; Mudgett, M.B.; Sattely, E.S. Arabidopsis UGT76b1 glycosylates N-hydroxy-pipecolic acid and inactivates systemic acquired resistance in tomato. Plant Cell 2021, 33, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Nothias, L.-F.; Vining, O.; Meehan, M.; Esquenazi, E.; Dorrestein, P.C. Molecular networking as a drug discovery, drug metabolism, and precision medicine strategy. Trends Pharmacol. Sci. 2017, 38, 143–154. [Google Scholar] [CrossRef]
- Li, D.; Gaquerel, E. Next-generation mass spectrometry metabolomics revives the functional analysis of plant metabolic diversity. Annu. Rev. Plant Biol. 2021, 72, 867–891. [Google Scholar] [CrossRef]
- Owen, C.; Patron, N.J.; Huang, A.; Osbourn, A. Harnessing plant metabolic diversity. Curr. Opin. Chem. Biol. 2017, 40, 24–30. [Google Scholar] [CrossRef]
- Polturak, G.; Osbourn, A. The emerging role of biosynthetic gene clusters in plant defense and plant interactions. PLoS Pathog. 2021, 17, e1009698. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. Pacbio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Bayega, A.; Fahiminiya, S.; Oikonomopoulos, S.; Ragoussis, J. Current and future methods for mRNA analysis: A drive toward single molecule sequencing. In Gene Expression Analysis: Methods and Protocols; Raghavachari, N., Garcia-Reyero, N., Eds.; Springer: New York, NY, USA, 2018; pp. 209–241. [Google Scholar]
- Michael, T.P.; VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of Pacbio and nanopore-based rna sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef]
- Altschuler, S.J.; Wu, L.F. Cellular heterogeneity: Do differences make a difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.; Tian, X.; Xu, J. Single-cell transcriptome analysis in plants: Advances and challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef] [PubMed]
- de Souza, L.P.; Borghi, M.; Fernie, A. Plant single-cell metabolomics-challenges and perspectives. Int. J. Mol. Sci. 2020, 21, 8987. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. Metlin: A technology platform for identifying knowns and unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. Pubchem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Pomyen, Y.; Wanichthanarak, K.; Poungsombat, P.; Fahrmann, J.; Grapov, D.; Khoomrung, S. Deep metabolome: Applications of deep learning in metabolomics. Comput. Struct. Biotechnol. J. 2020, 18, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Phelan, V.V. Feature-based molecular networking for metabolite annotation. Methods Mol. Biol. 2020, 2104, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.D.; Wang, D. Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 2020, 16, e1007677. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, F.; Yu, Z.; Zhou, Q.; Huang, A. Multi-Omics-Based Discovery of Plant Signaling Molecules. Metabolites 2022, 12, 76. https://doi.org/10.3390/metabo12010076
Luo F, Yu Z, Zhou Q, Huang A. Multi-Omics-Based Discovery of Plant Signaling Molecules. Metabolites. 2022; 12(1):76. https://doi.org/10.3390/metabo12010076
Chicago/Turabian StyleLuo, Fei, Zongjun Yu, Qian Zhou, and Ancheng Huang. 2022. "Multi-Omics-Based Discovery of Plant Signaling Molecules" Metabolites 12, no. 1: 76. https://doi.org/10.3390/metabo12010076
APA StyleLuo, F., Yu, Z., Zhou, Q., & Huang, A. (2022). Multi-Omics-Based Discovery of Plant Signaling Molecules. Metabolites, 12(1), 76. https://doi.org/10.3390/metabo12010076