
Comprehensive Biotransformation Analysis of Phenylalanine-Tyrosine Metabolism Reveals Alternative Routes of Metabolite Clearance in Nitisinone-Treated Alkaptonuria

 ,
,  ,
,  , , ,
, , ,
Abstract
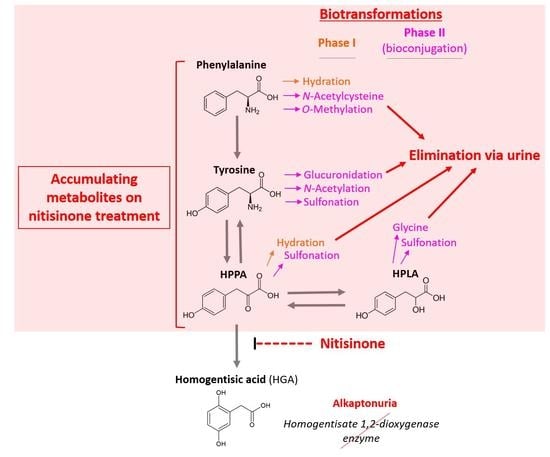
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Study Design and Patients
2.3. Sample Collection and Preparation
2.4. Quantitative Metabolite Data
2.5. LC-QTOF-MS Analysis
2.6. Data Pre-Processing and Statistical Analysis
3. Results
3.1. Summary of Metabolites Identified and Retained Post-QC Filtering
3.2. Effect of Nitisinone Treatment on Serum and Urine Metabolites
3.3. Temporal Changes in Metabolites over Long-Term Nitisinone Treatment
3.4. Effect of Nitisinone Treatment on Summed Metabolites
3.5. Correlation of Urine Metabolites against Quantitative Biochemical Data
3.6. Sex Differences in Metabolite Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthews, D.E. An Overview of Phenylalanine and Tyrosine Kinetics in Humans. J. Nutr. 2007, 137, 1549S. [Google Scholar] [CrossRef] [PubMed]
- Al Hafid, N.; Christodoulou, J. Phenylketonuria: A Review of Current and Future Treatments. Transl. Pediatr. 2015, 4, 304–317. [Google Scholar] [CrossRef]
- Holme, E.; Lindstedt, S. Tyrosinaemia Type I and NTBC (2-(2-Nitro-4-Trifluoromethylbenzoyl)-1,3-Cyclohexanedione). J. Inherit. Metab. Dis. 1998, 21, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, L.; Norman, B.; Gallagher, J. Ochronotic Pigmentation Is Caused by Homogentisic Acid and Is the Key Event in Alkaptonuria Leading to the Destructive Consequences of the Disease—A Review. J. Inherit. Metab. Dis. 2019, 42, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, S.; Holme, E.; Lock, E.A.; Hjalmarson, O.; Strandvik, B. Treatment of Hereditary Tyrosinaemia Type I by Inhibition of 4-Hydroxyphenylpyruvate Dioxygenase. Lancet 1992, 340, 813–817. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.T.; Dutton, J.J.; Fitzgerald, R.; Briggs, M.C.; Bygott, H.; Psarelli, E.E.; Cox, T.F.; Gallagher, J.A.; et al. Suitability of Nitisinone In Alkaptonuria 1 (SONIA 1): An International, Multicentre, Randomised, Open-Label, No-Treatment Controlled, Parallel-Group, Dose-Response Study to Investigate the Effect of Once Daily Nitisinone on 24-h Urinary Homogentisic Acid. Ann. Rheum. Dis. 2016, 75, 362–367. [Google Scholar] [CrossRef]
- Introne, W.J.; Perry, M.B.; Troendle, J.; Tsilou, E.; Kayser, M.A.; Suwannarat, P.; O’Brien, K.E.; Bryant, J.; Sachdev, V.; Reynolds, J.C.; et al. A 3-Year Randomized Therapeutic Trial of Nitisinone in Alkaptonuria. Mol. Genet. Metab. 2011, 103, 307–314. [Google Scholar] [CrossRef]
- Suwannarat, P.; O’Brien, K.; Perry, M.B.; Sebring, N.; Bernardini, I.; Kaiser-Kupfer, M.I.; Rubin, B.I.; Tsilou, E.; Gerber, L.H.; Gahl, W.A. Use of Nitisinone in Patients with Alkaptonuria. Metabolism 2005, 54, 719–728. [Google Scholar] [CrossRef]
- Phornphutkul, C.; Introne, W.J.; Perry, M.B.; Bernardini, I.; Murphey, M.D.; Fitzpatrick, D.L.; Anderson, P.D.; Huizing, M.; Anikster, Y.; Gerber, L.H.; et al. Natural History of Alkaptonuria. N. Engl. J. Med. 2002, 347, 2111–2121. [Google Scholar] [CrossRef]
- Milan, A.M.; Hughes, A.T.; Davison, A.S.; Devine, J.; Usher, J.; Curtis, S.; Khedr, M.; Gallagher, J.A.; Ranganath, L.R. The Effect of Nitisinone on Homogentisic Acid and Tyrosine: A Two-Year Survey of Patients Attending the National Alkaptonuria Centre, Liverpool. Ann. Clin. Biochem. 2017, 54, 323–330. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Couce, M.L.; Das, A.M.; de Laet, C.; Dionisi-Vici, C.; Lund, A.M.; Schiff, M.; Spada, M.; Sparve, E.; Szamosi, J.; et al. Long-Term Safety and Outcomes in Hereditary Tyrosinaemia Type 1 with Nitisinone Treatment: A 15-Year Non-Interventional, Multicentre Study. Lancet Diabetes Endocrinol. 2021, 9, 427–435. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Psarelli, E.E.; Arnoux, J.B.; Braconi, D.; Briggs, M.; Bröijersén, A.; Loftus, N.; Bygott, H.; Cox, T.F.; Davison, A.S.; et al. Efficacy and Safety of Once-Daily Nitisinone for Patients with Alkaptonuria (SONIA 2): An International, Multicentre, Open-Label, Randomised Controlled Trial. Lancet Diabetes Endocrinol. 2020, 8, 762–772. [Google Scholar] [CrossRef]
- Sloboda, N.; Wiedemann, A.; Merten, M.; Alqhatani, A.; Jeannesson, E.; Blum, A.; Henn-Ménétré, S.; Guéant, J.L.; Renard, E.; Feillet, F. Efficacy of Low Dose Nitisinone in the Management of Alkaptonuria. Mol. Genet. Metab. 2019, 127, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Khedr, M.; Cooper, M.S.; Hughes, A.T.; Milan, A.M.; Davison, A.S.; Norman, B.P.; Sutherland, H.; Jarvis, J.C.; Fitzgerald, R.; Markinson, L.; et al. Nitisinone Causes Acquired Tyrosinosis in Alkaptonuria. J. Inherit. Metab. Dis. 2020, 43, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.S.; Norman, B.; Milan, A.M.; Hughes, A.T.; Khedr, M.; Rovensky, J.; Gallagher, J.A.; Ranganath, L.R. Assessment of the Effect of Once Daily Nitisinone Therapy on 24-h Urinary Metadrenalines and 5-Hydroxyindole Acetic Acid Excretion in Patients with Alkaptonuria After 4 Weeks of Treatment. JIMD Rep. 2017, 41, 1–10. [Google Scholar] [CrossRef]
- Thimm, E.; Herebian, D.; Assmann, B.; Klee, D.; Mayatepek, E.; Spiekerkoetter, U. Increase of CSF Tyrosine and Impaired Serotonin Turnover in Tyrosinemia Type I. Mol. Genet. Metab. 2011, 102, 122–125. [Google Scholar] [CrossRef]
- Davison, A.S.; Strittmatter, N.; Sutherland, H.; Hughes, A.T.; Hughes, J.; Bou-Gharios, G.; Milan, A.M.; Goodwin, R.J.A.; Ranganath, L.R.; Gallagher, J.A. Assessing the Effect of Nitisinone Induced Hypertyrosinaemia on Monoamine Neurotransmitters in Brain Tissue from a Murine Model of Alkaptonuria Using Mass Spectrometry Imaging. Metabolomics 2019, 15, 68. [Google Scholar] [CrossRef]
- Zubarioglu, T. Nitisinone: A Review. Orphan Drugs Res. Rev. 2017, 7, 25–35. [Google Scholar]
- Davison, A.S.; Norman, B.P.; Ross, G.A.; Hughes, A.T.; Khedr, M.; Milan, A.M.; Gallagher, J.A.; Ranganath, L.R. Evaluation of the Serum Metabolome of Patients with Alkaptonuria before and after Two Years of Treatment with Nitisinone Using LC-QTOF-MS. JIMD Rep. 2019, 48, 67–74. [Google Scholar] [CrossRef]
- Khedr, M.; Judd, S.; Briggs, M.C.; Hughes, A.T.; Milan, A.M.; Stewart, R.M.K.; Lock, E.A.; Gallagher, J.A.; Ranganath, L.R. Asymptomatic Corneal Keratopathy Secondary to Hypertyrosinaemia Following Low Dose Nitisinone and a Literature Review of Tyrosine Keratopathy in Alkaptonuria. JIMD Rep. 2018, 40, 31–37. [Google Scholar] [CrossRef]
- Stewart, R.M.K.; Briggs, M.C.; Jarvis, J.C.; Gallagher, J.A.; Ranganath, L. Reversible Keratopathy Due to Hypertyrosinaemia Following Intermittent Low-Dose Nitisinone in Alkaptonuria: A Case Report. JIMD Rep. 2014, 17, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, B.P.; Davison, A.S.; Ross, G.A.; Milan, A.M.; Hughes, A.T.; Sutherland, H.; Jarvis, J.C.; Roberts, N.B.; Gallagher, J.A.; Ranganath, L.R. A Comprehensive LC-QTOF-MS Metabolic Phenotyping Strategy: Application to Alkaptonuria. Clin. Chem. 2019, 65, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Norman, B.P.; Davison, A.S.; Hughes, J.H.; Sutherland, H.; Wilson, P.J.; Berry, N.G.; Hughes, A.T.; Milan, A.M.; Jarvis, J.C.; Roberts, N.B.; et al. Metabolomic Studies in the Inborn Error of Metabolism Alkaptonuria Reveal New Biotransformations in Tyrosine Metabolism. Genes Dis. 2021, 9, 1129–1142. [Google Scholar] [CrossRef]
- Sandritter, T.; Jones, B.; GL, K.; Lowry, J. Principles of Drug Therapy. In Textbook of Pediatrics; Kliegman, R., St. Geme, J., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 445–456. [Google Scholar]
- Ranganath, L.R.; Hughes, A.T.; Davison, A.S.; Khedr, M.; Olsson, B.; Rudebeck, M.; Imrich, R.; Norman, B.P.; Bou-Gharios, G.; Gallagher, J.A.; et al. Temporal Adaptations in the Phenylalanine/Tyrosine Pathway and Related Factors during Nitisinone-Induced Tyrosinaemia in Alkaptonuria. Mol. Genet. Metab. 2022, in press. [Google Scholar] [CrossRef]
- Hughes, A.T.; Milan, A.M.; Davison, A.S.; Christensen, P.; Ross, G.; Gallagher, J.A.; Dutton, J.J.; Ranganath, L.R. Serum Markers in Alkaptonuria: Simultaneous Analysis of Homogentisic Acid, Tyrosine and Nitisinone by Liquid Chromatography Tandem Mass Spectrometry. Ann. Clin. Biochem. 2015, 52, 597–605. [Google Scholar] [CrossRef]
- Vorkas, P.A.; Isaac, G.; Anwar, M.A.; Davies, A.H.; Want, E.J.; Nicholson, J.K.; Holmes, E. Untargeted UPLC-MS Profiling Pipeline to Expand Tissue Metabolome Coverage: Application to Cardiovascular Disease. Anal. Chem. 2015, 87, 4184–4193. [Google Scholar] [CrossRef]
- Norman, B.; Davison, A.; Ross, G.A.; Milan, A.M.; Hughes, A.T.; Sutherland, H.; Jarvis, J.C.; Roberts, N.B.; Gallagher, J.A.; Ranganath, L. Three Accurate Mass Retention Time (AMRT) Databases Generated from IROA Technologies Metabolite Library of Standards by LC-QTOF-MS Analysis. Figshare. Collection. 2019. [Google Scholar]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed Minimum Reporting Standards for Chemical Analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.; Davison, A.S.; Khedr, M.; Norman, B.P.; Bou-Gharios, G.; Gallagher, J.A.; Imrich, R.; Arnoux, J.B.; et al. Determinants of Tyrosinaemia during Nitisinone Therapy in Alkaptonuria. Sci. Rep. 2022, 12, 16083. [Google Scholar] [CrossRef]
- Williams, D.P.; Lawrence, A.; Meng, X. Pharmacological and Toxicological Considerations of Homogentisic Acid in Alkaptonuria. Pharmacologia 2012, 3, 61–74. [Google Scholar] [CrossRef]
- Testa, B.; van de Waterbeemd, H. ADME-Tox Approaches. In Comprehensive Medicinal Chemistry II; Taylor, J., Triggle, D., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2007; Volume 5. [Google Scholar]
- De Kanter, R.; De Jager, M.H.; Draaisma, A.L.; Jurva, J.U.; Olinga, P.; Meijer, D.K.F.; Groothuis, G.M.M. Drug-Metabolizing Activity of Human and Rat Liver, Lung, Kidney and Intestine Slices. Xenobiotica 2002, 32, 349–362. [Google Scholar] [CrossRef]
- Gertsman, I.; Barshop, B.A.; Panyard-Davis, J.; Gangoiti, J.A.; Nyhan, W.L. Metabolic Effects of Increasing Doses of Nitisinone in the Treatment of Alkaptonuria. JIMD Reports 2015, 24, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Hodgson, E. Metabolism of Toxicants. In A Textbook of Modern Toxicology, 3rd ed.; Hodgson, E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human Sulfotransferases and Their Role in Chemical Metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef]
- Tallan, H.H.; Bella, S.T.; Stein, W.H.; Moore, S. Tyrosine-O-Sulfate as a Constituent of Normal Human Urine. J. Biol. Chem. 1955, 217, 703–708. [Google Scholar] [CrossRef]
- Hext, P.M.; Thomas, S.; Rose, F.A.; Dodgson, K.S. Determination and Significance of L-Tyrosine O-Sulphate and Its Deaminated Metabolites in Normal Human and Mouse Urine. Biochem. J. 1973, 134, 629. [Google Scholar] [CrossRef]
- John, R.A.; Rose, F.A.; Wusteman, F.S.; Dodgson, K.S. The Detection and Determination of L-Tyrosine O-Sulphate in Rabbit and Other Mammalian Urine. Biochem. J. 1966, 100, 278. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Takami, Y.; Zwieb, C.; Nakayama, T.; Suiko, M.; Nakajima, H.; Liu, M.C. Purification, Characterization, and Molecular Cloning of a Novel Rat Liver Dopa/Tyrosine Sulfotransferase. J. Biol. Chem. 1995, 270, 30470–30478. [Google Scholar] [CrossRef]
- Yang, Y.S.; Wang, C.C.; Chen, B.H.; Hou, Y.H.; Hung, K.S.; Mao, Y.C. Tyrosine Sulfation as a Protein Post-Translational Modification. Molecules 2015, 20, 2138–2164. [Google Scholar] [CrossRef] [PubMed]
- Pencharz, P.B.; Hsu, J.W.C.; Ball, R.O. Aromatic Amino Acid Requirements in Healthy Human Subjects. J. Nutr. 2007, 137, 1576S–1578S. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P.F.; Malgarin, F.; Cararo, J.H.; Cardoso, F.; Streck, E.L.; Ferreira, G.C. Phenylketonuria Pathophysiology: On the Role of Metabolic Alterations. Aging Dis. 2015, 6, 390–399. [Google Scholar] [CrossRef]
- Williams, R.A.; Mamotte, C.D.; Burnett, J.R.; Prof, C.; Burnett, J. Phenylketonuria: An Inborn Error of Phenylalanine Metabolism. Clin. Biochem. Rev. 2008, 29, 31. [Google Scholar]
- Donlon, J.; Sarkissian, C.; Levy, H.; Scriver, C. Hyperphenylalaninemia: Phenylalanine Hydroxylase Deficiency. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D., Antonarakis, S., Ballabio, A., Beaudet, A., Mitchell, G., Eds.; McGraw Hill: New York, NY, USA, 2019. [Google Scholar]
- Gissen, P.; Preece, M.A.; Willshaw, H.A.; McKiernan, P.J. Ophthalmic Follow-up of Patients with Tyrosinaemia Type I on NTBC. J. Inherit. Metab. Dis. 2003, 26, 13–16. [Google Scholar] [CrossRef]
- Ranganath, L.; Milan, A.; Hughes, A.; Davison, A.; Khedr, M.; Imrich, R.; Arnoux, J.; Rudebeck, M.; Olsson, B.; Norman, B.; et al. Comparing the Phenylalanine/Tyrosine Pathway and Related Factors between Keratopathy and No-Keratopathy Groups as Well as between Men and Women in Alkaptonuria Patients Receiving Nitisinone. Metabolites 2022, 12, 772. [Google Scholar] [CrossRef] [PubMed]
- Milan, A.M.; Hughes, A.T.; Davison, A.S.; Khedr, M.; Rovensky, J.; Psarelli, E.E.; Cox, T.F.; Rhodes, N.P.; Gallagher, J.A.; Ranganath, L.R. Quantification of the Flux of Tyrosine Pathway Metabolites during Nitisinone Treatment of Alkaptonuria. Sci. Rep. 2019, 9, 10024. [Google Scholar] [CrossRef]
- Chow, W.Y.; Norman, B.P.; Roberts, N.B.; Ranganath, L.R.; Teutloff, C.; Bittl, R.; Duer, M.J.; Gallagher, J.A.; Oschkinat, H. Pigmentation Chemistry and Radical-Based Collagen Degradation in Alkaptonuria and Osteoarthritic Cartilage. Angew. Chem. Int. Ed. Engl. 2020, 59, 11937–11942. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for Large-Scale Metabolic Profiling of Serum and Plasma Using Gas Chromatography and Liquid Chromatography Coupled to Mass Spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Urine | Serum | |||
|---|---|---|---|---|
| Untreated | Treated | Untreated | Treated | |
| Mean age (SD) | 46.7 (9.8) | 47.8 (11.2) | 47.0 (9.9) | 46.7 (11.3) |
| Total male | 28 | 37 | 25 | 34 |
| Total female | 22 | 16 | 20 | 13 |
| Total Asian | 2 | 1 | 1 | 1 |
| Total black | 0 | 1 | 0 | 0 |
| Total Caucasian | 48 | 51 | 45 | 45 |
| Numbers of patients by site | ||||
| Paris | 12 | 15 | 8 | 11 |
| Liverpool | 14 | 14 | 14 | 13 |
| Piešťany | 24 | 24 | 23 | 23 |
| Urine | Serum | ||||||
|---|---|---|---|---|---|---|---|
| Metabolite | Formula | Theoretical Neutral Monoisotopic Mass | Retention Time (RT) | Preferred Polarity | Basis for ID | Preferred Polarity | Basis for ID |
| Phenylalanine | C9H11NO2 | 165.0795 | 3.7 | (+) | AM, MS2, RT | (+) | AM, MS2, RT |
| Phenylalanine hydrate | C9H13NO3 | 183.0869 | 2.2 | (+) | AM, MS2 ** | ||
| Phenylalanine N-acetylcysteine | C14H18N2O5 S | 326.0966 | 4.7 | (−) | AM, MS2 ** | ||
| O-Methyl-phenylalanine | C10H13NO2 | 179.0942 | 4.9 | (−) | AM, MS2 ** | ||
| Phenylpyruvic acid | C9H8O3 | 164.0472 | 4.7 | (+) | AM, MS2 ** | ||
| Phenyllactic acid | C9H10O3 | 166.063 | 6.7 | (−) | AM, MS2 ** | ||
| Phenylacetamide | C8H9NO | 135.0685 | 4.8 | (+) | AM, MS2 | ||
| Phenylacetylglutamine | C13H16N2O4 | 264.1114 | 5.5 | (+) | AM, MS2 ** | (+) | AM, MS2 ** |
| Tyrosine | C9H11NO3 | 181.0756 | 2.2 | (−) | AM, MS2, RT | (+) | AM, MS2, RT |
| N-Acetyl-tyrosine | C11H13NO4 | 223.085 | 4.9 | (−) | AM, MS2, RT | (+) | AM, MS2, RT |
| Tyrosine-sulfate | C9H11NO6 S | 261.0306 | 2.0 | (−) | AM, MS2 ** | ||
| Tyrosine-glucuronide | C15H19NO9 | 357.1009 | 3.9 | (+) | AM, MS2 ** | ||
| Tyramine * | C8H11NO | 137.0841 | 2.4 | (+) | AM, MS2, RT | ||
| HPPA | C9H8O4 | 180.0425 | 3.7 | (−) | AM, MS2, RT | (−) | AM, RT |
| HPPA-hydrate | C9H10O5 | 198.053 | 3.6 | (+) | AM, MS2 ** | ||
| HPPA-sulfate | C9H8O7S | 259.9991 | 5.3 | (−) | AM, MS2 ** | ||
| HPLA | C9H10O4 | 182.058 | 4.7 | (−) | AM, MS2, RT | (−) | AM, MS2, RT |
| HPLA-glycine | C11H13NO5 | 239.0774 | 4.7 | (−) | AM, MS2 | ||
| HPLA-sulfate | C9H10O7S | 262.0146 | 3.8 | (−) | AM, MS2 ** | ||
| HGA | C8H8O4 | 168.0425 | 3.5 | (−) | AM, MS2, RT | (−) | AM, MS2, RT |
| Acetyl-HGA | C10H10O5 | 210.0525 | 6.4 | (−) | AM, MS2 ** | ||
| HGA-glucuronide | C14H16 O10 | 344.0737 | 2.5 | (+) | AM, MS2 ** | ||
| HGA-sulfate | C8H8O7S | 247.9991 | 2.8 | (−) | AM, MS2 ** | (+) | AM, MS2 ** |
| Urine | Serum | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treated | Untreated | Treated | Untreated | |||||||||
| Metabolite | p-value (adjusted) | V4 vs. V1 | V6 vs. V1 | p-value (adjusted) | V4 vs. V1 | V6 vs. V1 | p-value (adjusted) | V4 vs. V1 | V6 vs. V1 | p-value (adjusted) | V4 vs. V1 | V6 vs. V1 |
| Phenylalanine | <0.0001 | 1.2 ↑ | 1.7 ↑ | <0.0001 | 1.0 ↓ | 1.4 ↑ | <0.001 | 1.0 ↑ | 1.3 ↑ | 0.0094 | 1.2 ↑ | 1.3 ↑ |
| Phenylalanine hydrate | <0.0001 | 2.2 ↑ | 3.2 ↑ | <0.001 | 1.0 ↓ | 1.3 ↑ | ||||||
| Phenylalanine- N-acetylcysteine | <0.0001 | 9.3 ↑ | 10.6 ↑ | <0.0001 | 1.4 ↑ | 1.8 ↑ | ||||||
| O-Methyl-phenylalanine | <0.0001 | 5.5 ↑ | 6.4 ↑ | 0.062 (NS) | 1.1 ↓ | 1.2 ↑ | ||||||
| Phenylpyruvic acid | <0.0001 | 11.4 ↑ | 12.6 ↑ | <0.001 | 1.0 ↑ | 1.2 ↑ | ||||||
| Phenyllactic acid | <0.0001 | 4.9 ↑ | 6.0 ↑ | 0.0063 | 1.1 ↑ | 1.3 ↑ | ||||||
| Phenylacetamide | <0.0001 | 4.1 ↑ | 5.2 ↑ | <0.0001 | 1.1 ↓ | 1.3 ↑ | ||||||
| Phenylacetylglutamine | <0.0001 | 1.2 ↑ | 1.6 ↑ | <0.0001 | 1.0 ↑ | 1.5 ↑ | <0.0001 | 1.3 ↑ | 1.7 ↑ | 0.0094 | 1.3 ↑ | 1.4 ↑ |
| Tyrosine | <0.0001 | 4.0 ↑ | 4.7 ↑ | <0.001 | 1.1 ↓ | 1.2 ↑ | <0.0001 | 5.4 ↑ | 5.7 ↑ | <0.0001 | 1.4 ↑ | 1.4 ↑ |
| N-Acetyl-tyrosine | <0.0001 | 5.7 ↑ | 6.8 ↑ | <0.0001 | 1.1 ↓ | 1.2 ↑ | <0.0001 | 4.3 ↑ | 5.4 ↑ | 0.0022 | 1.1 ↑ | 1.2 ↑ |
| Tyrosine-sulfate | <0.0001 | 1.6 ↑ | 1.9 ↑ | <0.0001 | 1.0 ↓ | 1.4 ↑ | ||||||
| Tyrosine-glucuronide | <0.0001 | 8.0 ↑ | 7.9 ↑ | 0.018 | 1.3 ↑ | 1.4 ↑ | ||||||
| Tyramine | 0.0028 | 2.1 ↑ | 1.2 ↑ | 0.018 | 1.1 ↓ | 1.3 ↑ | ||||||
| HPPA | <0.0001 | 20.0 ↑ | 22.7 ↑ | <0.001 | 1.1 ↓ | 1.1 ↑ | <0.0001 | 14.3 ↑ | 15.0 ↑ | <0.0001 | 1.4 ↑ | 1.5 ↑ |
| HPPA-hydrate | <0.0001 | 2.4 ↑ | 3.6 ↑ | <0.0001 | 1.0 ↓ | 1.5 ↑ | ||||||
| HPPA-sulfate | <0.0001 | 1.7 ↑ | 2.6 ↑ | <0.001 | 1.3 ↑ | 1.6 ↑ | ||||||
| HPLA | <0.0001 | 9.4 ↑ | 10.6 ↑ | 0.0063 | 1.0 ↑ | 1.3 ↑ | <0.0001 | 12.8 ↑ | 13.3 ↑ | 0.19 (NS) | 1.1 ↑ | 1.1 ↑ |
| HPLA-glycine | <0.0001 | 16.2 ↑ | 15.2 ↑ | <0.0001 | 1.0 ↓ | 1.2 ↑ | ||||||
| HPLA-sulfate | <0.0001 | 6.4 ↑ | 7.9 ↑ | <0.0001 | 1.1 ↑ | 1.4 ↑ | ||||||
| HGA | <0.0001 | 10.9 ↓ | 8.0 ↓ | <0.0001 | 1.0 ↓ | 1.2 ↑ | <0.0001 | 12.0 ↓ | 10.2 ↓ | 0.0094 | 1.0 ↑ | 1.3 ↑ |
| Acetyl-HGA | <0.0001 | 4.0 ↓ | 2.7 ↓ | 0.011 | 1.0 ↓ | 1.3 ↑ | ||||||
| HGA-glucuronide | <0.0001 | 18.2 ↓ | 20.5 ↓ | <0.001 | 1.1 ↓ | 1.2 ↑ | ||||||
| HGA-sulfate | <0.0001 | 18.4 ↓ | 12.1 ↓ | <0.001 | 1.0 ↓ | 1.2 ↑ | <0.001 | 1.9 ↓ | 1.3 ↓ | <0.0001 | 2.7 ↑ | 2.9 ↑ |
| Untreated | Treated | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit 1 | Visit 4 | Visit 6 | Visit 1 | Visit 4 | Visit 6 | |||||||
| Abundance | Contribution (%) | Abundance | Contribution (%) | Abundance | Contribution (%) | Abundance | Contribution (%) | Abundance | Contribution (%) | Abundance | Contribution (%) | |
| Urine metabolites (grouped) | ||||||||||||
| Phenylalanine (+metabs) | 4.99 × 106 | 40.9 | 5.21 × 106 | 42.5 | 8.68 × 106 | 42.7 | 5.19 × 106 | 40.1 | 7.13 × 106 | 36.5 | 1.01 × 107 | 37.7 |
| Tyrosine (+metabs) | 5.85 × 104 | 0.5 | 5.16 × 104 | 0.5 | 8.83 × 104 | 0.5 | 6.71 × 104 | 0.5 | 7.59 × 105 | 3.8 | 9.37 × 105 | 3.4 |
| HPPA (+metabs) | 2.47 × 104 | 0.2 | 2.86 × 104 | 0.3 | 4.64 × 104 | 0.2 | 2.88 × 104 | 0.2 | 3.28 × 106 | 16.8 | 4.49 × 106 | 17.4 |
| HPLA (+metabs) | 1.20 × 105 | 1.0 | 1.39 × 105 | 1.2 | 2.42 × 105 | 1.1 | 1.19 × 105 | 1.0 | 7.26 × 106 | 39.3 | 9.91 × 106 | 37.3 |
| HGA (+metabs) | 6.51 × 106 | 57.4 | 6.26 × 106 | 55.7 | 1.02 × 107 | 55.4 | 6.97 × 106 | 58.2 | 4.01 × 105 | 3.6 | 6.94 × 105 | 4.3 |
| Total urine metabolites | 1.17 × 107 | - | 1.17 × 107 | - | 2.28 × 107 | - | 1.24 × 107 | - | 1.88 × 107 | - | 2.62 × 107 | - |
| Serum metabolites (grouped) | ||||||||||||
| Phenylalanine (+metabs) | 1.61 × 107 | 79.9 | 1.83 × 107 | 76.7 | 2.00 × 107 | 76.9 | 1.58 × 107 | 79.5 | 1.64 × 107 | 28.5 | 2.06 × 107 | 31.2 |
| Tyrosine (+metabs) | 3.25 × 106 | 16.3 | 4.81 × 106 | 20.4 | 5.14 × 106 | 19.3 | 3.19 × 106 | 16.3 | 3.53 × 107 | 56.5 | 3.85 × 107 | 53.7 |
| HPPA (+metabs) | 3.12 × 103 | 0.0 | 5.72 × 103 | 0.0 | 6.48 × 103 | 0.0 | 2.99 × 103 | 0.0 | 5.76 × 105 | 0.9 | 7.00 × 105 | 0.9 |
| HPLA (+metabs) | 5.24 × 104 | 0.4 | 5.52 × 104 | 0.3 | 5.83 × 104 | 0.2 | 4.90 × 104 | 0.3 | 9.00 × 106 | 14.0 | 1.04 × 107 | 13.9 |
| HGA (+metabs) | 6.38 × 105 | 3.5 | 6.09 × 105 | 2.6 | 9.98 × 105 | 3.6 | 7.28 × 105 | 3.8 | 3.91 × 104 | 0.2 | 7.25 × 104 | 0.3 |
| Total serum metabolites | 2.01 × 107 | - | 2.38 × 107 | - | 2.62 × 107 | - | 1.98 ×107 | - | 6.13 × 107 | - | 7.04 × 107 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norman, B.P.; Davison, A.S.; Hickton, B.; Ross, G.A.; Milan, A.M.; Hughes, A.T.; Wilson, P.J.M.; Sutherland, H.; Hughes, J.H.; Roberts, N.B.; et al. Comprehensive Biotransformation Analysis of Phenylalanine-Tyrosine Metabolism Reveals Alternative Routes of Metabolite Clearance in Nitisinone-Treated Alkaptonuria. Metabolites 2022, 12, 927. https://doi.org/10.3390/metabo12100927
Norman BP, Davison AS, Hickton B, Ross GA, Milan AM, Hughes AT, Wilson PJM, Sutherland H, Hughes JH, Roberts NB, et al. Comprehensive Biotransformation Analysis of Phenylalanine-Tyrosine Metabolism Reveals Alternative Routes of Metabolite Clearance in Nitisinone-Treated Alkaptonuria. Metabolites. 2022; 12(10):927. https://doi.org/10.3390/metabo12100927
Chicago/Turabian StyleNorman, Brendan P., Andrew S. Davison, Bryony Hickton, Gordon A. Ross, Anna M. Milan, Andrew T. Hughes, Peter J. M. Wilson, Hazel Sutherland, Juliette H. Hughes, Norman B. Roberts, and et al. 2022. "Comprehensive Biotransformation Analysis of Phenylalanine-Tyrosine Metabolism Reveals Alternative Routes of Metabolite Clearance in Nitisinone-Treated Alkaptonuria" Metabolites 12, no. 10: 927. https://doi.org/10.3390/metabo12100927
APA StyleNorman, B. P., Davison, A. S., Hickton, B., Ross, G. A., Milan, A. M., Hughes, A. T., Wilson, P. J. M., Sutherland, H., Hughes, J. H., Roberts, N. B., Bou-Gharios, G., Gallagher, J. A., & Ranganath, L. R. (2022). Comprehensive Biotransformation Analysis of Phenylalanine-Tyrosine Metabolism Reveals Alternative Routes of Metabolite Clearance in Nitisinone-Treated Alkaptonuria. Metabolites, 12(10), 927. https://doi.org/10.3390/metabo12100927