
COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency



{kind=link}
Abstract
:1. Introduction
2. Case Description
3. Primary Coenzyme Q10 Deficiency-4
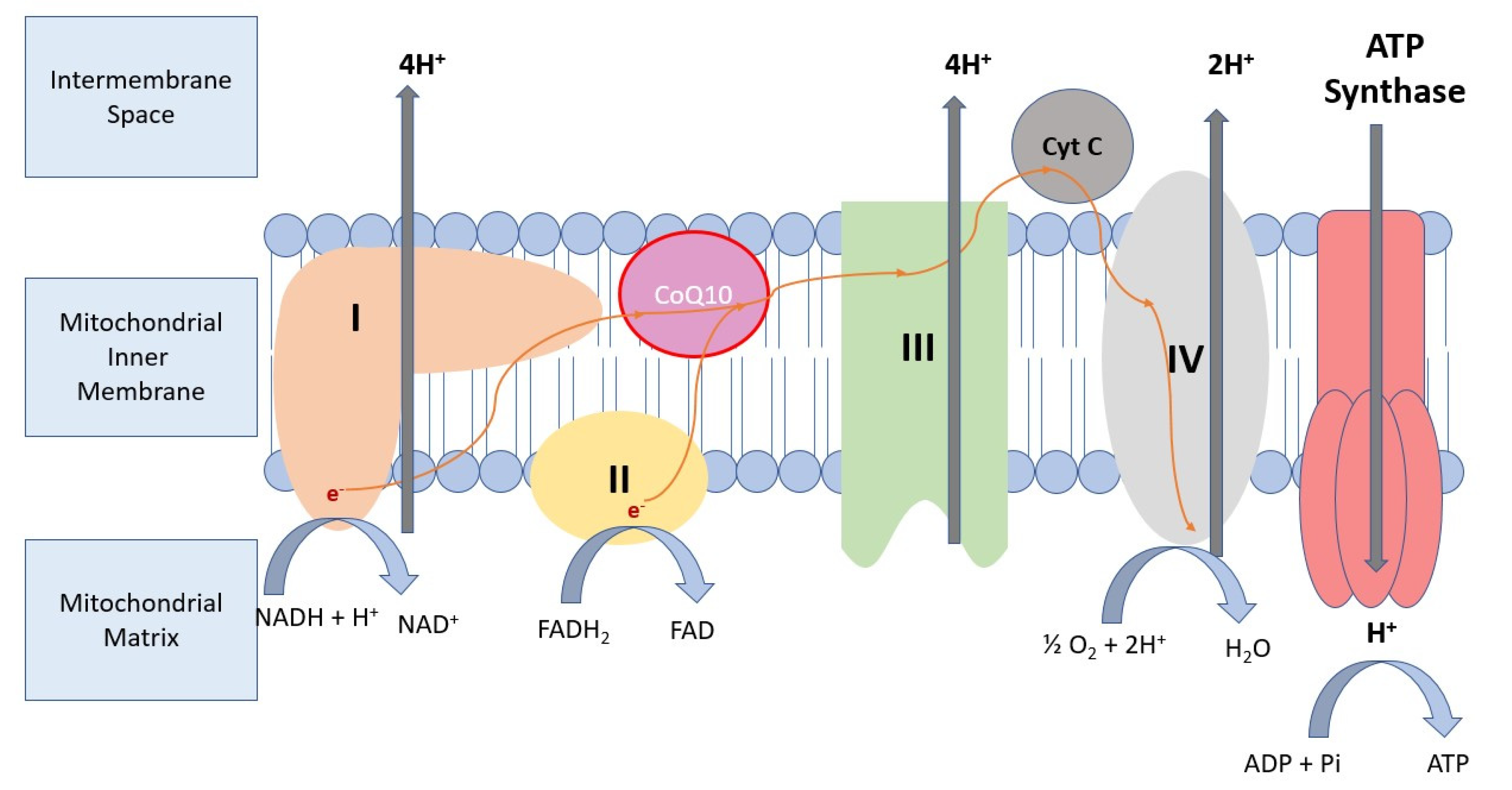
3.1. Molecular Background
3.2. Clinical Manifestations
3.3. Diagnosis
3.4. Treatment and Management
3.5. CoQ10 Pharmacokinetics
3.6. Outcomes
3.7. Approach to Further Therapies
4. Differential Diagnosis of Other Mitochondrial Disorders Presenting with Ataxia
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Musselman, K.E.; Stoyanov, C.T.; Marasigan, R.; Jenkins, M.E.; Konczak, J.; Morton, S.M.; Bastian, A.J. Prevalence of Ataxia in Children: A Systematic Review. Neurology 2014, 82, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traschütz, A.; Schirinzi, T.; Laugwitz, L.; Murray, N.H.; Bingman, C.A.; Reich, S.; Kern, J.; Heinzmann, A.; Vasco, G.; Bertini, E.; et al. Clinico-Genetic, Imaging and Molecular Delineation of COQ8A-Ataxia: A Multicenter Study of 59 Patients. Ann. Neurol. 2020, 88, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Laredj, L.N.; Licitra, F.; Puccio, H.M. The Molecular Genetics of Coenzyme Q Biosynthesis in Health and Disease. Biochimie 2014, 100, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Emmanuele, V.; López, L.C.; López, L.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; et al. Heterogeneity of Coenzyme Q10 Deficiency: Patient Study and Literature Review. Arch. Neurol. 2012, 69, 978–983. [Google Scholar] [CrossRef] [Green Version]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; de Lonlay, P.; Ogier de Baulny, H.; et al. CABC1 Gene Mutations Cause Ubiquinone Deficiency with Cerebellar Ataxia and Seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Mutlu-Albayrak, H.; Kırat, E.; Gürbüz, G. Childhood-Onset Autosomal Recessive Ataxias: A Cross-Sectional Study from Turkey. Neurogenetics 2020, 21, 59–66. [Google Scholar] [CrossRef]
- Artuch, R.; Brea-Calvo, G.; Briones, P.; Aracil, A.; Galván, M.; Espinós, C.; Corral, J.; Volpini, V.; Ribes, A.; Andreu, A.L.; et al. Cerebellar Ataxia with Coenzyme Q10 Deficiency: Diagnosis and Follow-up after Coenzyme Q10 Supplementation. J. Neurol. Sci. 2006, 246, 153–158. [Google Scholar] [CrossRef]
- Değerliyurt, A.; Gülleroğlu, N.B.; Kibar Gül, A.E. Primary CoQ 10 Deficiency with a Severe Phenotype Due to the c.901 C > T (p.R301W) Mutation in the COQ8A Gene. Int. J. Neurosci. 2022, 4, 1–5. [Google Scholar] [CrossRef]
- Jacobsen, J.C.; Whitford, W.; Swan, B.; Taylor, J.; Love, D.R.; Hill, R.; Molyneux, S.; George, P.M.; Mackay, R.; Robertson, S.P.; et al. Compound Heterozygous Inheritance of Mutations in Coenzyme Q8A Results in Autosomal Recessive Cerebellar Ataxia and Coenzyme Q10 Deficiency in a Female Sib-Pair. JIMD Rep. 2018, 42, 31–36. [Google Scholar] [CrossRef]
- Malgireddy, K.; Thompson, R.; Torres-Russotto, D. A Novel CABC1/ADCK3 Mutation in Adult-Onset Cerebellar Ataxia. Park. Relat. Disord. 2016, 33, 151–152. [Google Scholar] [CrossRef]
- Shalata, A.; Edery, M.; Habib, C.; Genizi, J.; Mahroum, M.; Khalaily, L.; Assaf, N.; Segal, I.; Abed El Rahim, H.; Shapira, H.; et al. Primary Coenzyme Q Deficiency Due to Novel ADCK3 Variants, Studies in Fibroblasts and Review of Literature. Neurochem. Res. 2019, 44, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK3 Mutations with Epilepsy, Stroke-like Episodes and Ataxia: A POLG Mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Barca, E.; Musumeci, O.; Montagnese, F.; Marino, S.; Granata, F.; Nunnari, D.; Peverelli, L.; DiMauro, S.; Quinzii, C.M.; Toscano, A. Cerebellar Ataxia and Severe Muscle CoQ10 Deficiency in a Patient with a Novel Mutation in ADCK3. Clin. Genet. 2016, 90, 156–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, M.R.; Haghighi, R.; Badv, R.S.; Ghabeli, H.; Tavasoli, A.R.; Pourbakhtyaran, E.; Rezaei, Z.; Mahdieh, N.; Mohammadi, P.; Heidari, M. Epilepsia Partialis Continua a Clinical Feature of a Missense Variant in the ADCK3 Gene and Poor Response to Therapy. J. Mol. Neurosci. 2022, 72, 1125–1132. [Google Scholar] [CrossRef]
- Schirinzi, T.; Favetta, M.; Romano, A.; Sancesario, A.; Summa, S.; Minosse, S.; Zanni, G.; Castelli, E.; Bertini, E.; Petrarca, M.; et al. One-Year Outcome of Coenzyme Q10 Supplementation in ADCK3 Ataxia (ARCA2). Cerebellum Ataxias 2019, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.; Ruiz-Lopez, M.; Slow, E.; Tarnopolsky, M.; Lang, A.E.; Munhoz, R.P. ADCK3-Related Coenzyme Q10 Deficiency: A Potentially Treatable Genetic Disease. Mov. Disord. Clin. Pract. 2018, 5, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-T.; Hersheson, J.; Plagnol, V.; Fawcett, K.; Duberley, K.E.C.; Preza, E.; Hargreaves, I.P.; Chalasani, A.; Laurá, M.; Wood, N.W.; et al. Autosomal-Recessive Cerebellar Ataxia Caused by a Novel ADCK3 Mutation That Elongates the Protein: Clinical, Genetic and Biochemical Characterisation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Blumkin, L.; Leshinsky-Silver, E.; Zerem, A.; Yosovich, K.; Lerman-Sagie, T.; Lev, D. Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. In JIMD Reports; Springer: Cham, Switzerland, 2013; Volume 12, pp. 103–107. [Google Scholar]
- Hajjari, M.; Tahmasebi-Birgani, M.; Mohammadi-asl, J.; Nasiri, H.; Kollaee, A.; Mahmoodi, M.; Ansari, H. Exome Sequencing Found a Novel Homozygous Deletion in ADCK3 Gene Involved in Autosomal Recessive Spinocerebellar Ataxia. Gene 2019, 708, 10–13. [Google Scholar] [CrossRef]
- Gerards, M.; van den Bosch, B.; Calis, C.; Schoonderwoerd, K.; van Engelen, K.; Tijssen, M.; de Coo, R.; van der Kooi, A.; Smeets, H. Nonsense Mutations in CABC1/ADCK3 Cause Progressive Cerebellar Ataxia and Atrophy. Mitochondrion 2010, 10, 510–515. [Google Scholar] [CrossRef]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted Exome Analysis Identifies the Genetic Basis of Disease in over 50% of Patients with a Wide Range of Ataxia-Related Phenotypes. Genet. Med. 2019, 21, 195–206. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Shao, Y.-R.; Dong, Y.; Dong, H.-L.; Yang, L.; Ma, Y.; Shen, Y.; Wu, Z.-Y. Genetic Spectrum and Clinical Features in a Cohort of Chinese Patients with Autosomal Recessive Cerebellar Ataxias. Transl. Neurodegener. 2021, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Amprosi, M.; Zech, M.; Steiger, R.; Nachbauer, W.; Eigentler, A.; Gizewski, E.R.; Guger, M.; Indelicato, E.; Boesch, S. Familial Writer’s Cramp: A Clinical Clue for Inherited Coenzyme Q10 Deficiency. Neurogenetics 2021, 22, 81–86. [Google Scholar] [CrossRef]
- Wirth, T.; Tranchant, C.; Drouot, N.; Keren, B.; Mignot, C.; Cif, L.; Lefaucheur, R.; Lion-François, L.; Méneret, A.; Gras, D.; et al. Increased Diagnostic Yield in Complex Dystonia through Exome Sequencing. Park. Relat. Disord. 2020, 74, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Zhou, Z.; Hu, Z.; Du, J.; Liao, X.; Luo, Y.; Wang, J.; Yan, X.; Jiang, H.; Tang, B.; et al. Homozygosity Mapping and next Generation Sequencing for the Genetic Diagnosis of Hereditary Ataxia and Spastic Paraplegia in Consanguineous Families. Park. Relat. Disord. 2020, 80, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Galosi, S.; Barca, E.; Carrozzo, R.; Schirinzi, T.; Quinzii, C.M.; Lieto, M.; Vasco, G.; Zanni, G.; Di Nottia, M.; Galatolo, D.; et al. Dystonia-Ataxia with Early Handwriting Deterioration in COQ8A Mutation Carriers: A Case Series and Literature Review. Park. Relat. Disord. 2019, 68, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Czermin, B.; Gulati, S.; Demuth, S.; Houge, G.; Pyle, A.; Dineiger, C.; Blakely, E.L.; Hassani, A.; Foley, C.; et al. Adult-Onset Cerebellar Ataxia Due to Mutations in CABC1/ADCK3. J. Neurol. Neurosurg. Psychiatry 2012, 83, 174–178. [Google Scholar] [CrossRef]
- Uccella, S.; Pisciotta, L.; Severino, M.; Bertini, E.; Giacomini, T.; Zanni, G.; Prato, G.; De Grandis, E.; Nobili, L.; Mancardi, M.M. Photoparoxysmal Response in ADCK3 Autosomal Recessive Ataxia: A Case Report and Literature Review. Epileptic Disord. 2021, 23, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an Ancestral Kinase, Is Mutated in a Form of Recessive Ataxia Associated with Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terracciano, A.; Renaldo, F.; Zanni, G.; D’Amico, A.; Pastore, A.; Barresi, S.; Valente, E.M.; Piemonte, F.; Tozzi, G.; Carrozzo, R.; et al. The Use of Muscle Biopsy in the Diagnosis of Undefined Ataxia with Cerebellar Atrophy in Children. Eur. J. Paediatr. Neurol. 2012, 16, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Krygier, M.; Kwarciany, M.; Wasilewska, K.; Pienkowski, V.M.; Krawczyńska, N.; Zielonka, D.; Kosińska, J.; Stawinski, P.; Rudzińska-Bar, M.; Boczarska-Jedynak, M.; et al. A Study in a Polish Ataxia Cohort Indicates Genetic Heterogeneity and Points to MTCL1 as a Novel Candidate Gene. Clin. Genet. 2019, 95, 415–419. [Google Scholar] [CrossRef]
- Mignot, C.; Apartis, E.; Durr, A.; Marques Lourenço, C.; Charles, P.; Devos, D.; Moreau, C.; de Lonlay, P.; Drouot, N.; Burglen, L.; et al. Phenotypic Variability in ARCA2 and Identification of a Core Ataxic Phenotype with Slow Progression. Orphanet J. Rare Dis. 2013, 8, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ashizawa, T.; Peng, D. Primary Coenzyme Q10 Deficiency Due to COQ8A Gene Mutations. Mol. Genet. Genom. Med. 2020, 8, e1420. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ma, D.; Li, J.; Luo, C.; Sun, Y.; Zhang, J.; Hu, P.; Tang, W.; Xu, Z. A Novel COQ8A Missense Variant Associated with a Mild Form of Primary Coenzyme Q10 Deficiency Type 4. Clin. Biochem. 2020, 84, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Cotta, A.; Alston, C.L.; Baptista-Junior, S.; Paim, J.F.; Carvalho, E.; Navarro, M.M.; Appleton, M.; Ng, Y.S.; Valicek, J.; Da-Cunha-Junior, A.L.; et al. Early-Onset Coenzyme Q10 Deficiency Associated with Ataxia and Respiratory Chain Dysfunction Due to Novel Pathogenic COQ8A Variants, Including a Large Intragenic Deletion. JIMD Rep. 2020, 54, 45–53. [Google Scholar] [CrossRef]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.-L.; Davoine, C.-S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurol. 2018, 75, 591–599. [Google Scholar] [CrossRef]
- Nair, P.; Lama, M.; El-Hayek, S.; Abou Sleymane, G.; Stora, S.; Obeid, M.; Al-Ali, M.T.; Delague, V.; Mégarbané, A. COQ8A and MED25 Mutations in a Child with Intellectual Disability, Microcephaly, Seizures, and Spastic Ataxia: Synergistic Effect of Digenic Variants? Mol. Syndromol. 2019, 9, 319–323. [Google Scholar] [CrossRef]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A.M. Mitochondrial Disease Criteria: Diagnostic Applications in Children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Stefely, J.A.; Reidenbach, A.G.; Ulbrich, A.; Oruganty, K.; Floyd, B.J.; Jochem, A.; Saunders, J.M.; Johnson, I.E.; Minogue, C.E.; Wrobel, R.L.; et al. Mitochondrial ADCK3 Employs an Atypical Protein Kinase-like Fold to Enable Coenzyme Q Biosynthesis. Mol. Cell 2015, 57, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Oruganty, K.; Talevich, E.E.; Neuwald, A.F.; Kannan, N. Identification and Classification of Small Molecule Kinases: Insights into Substrate Recognition and Specificity. BMC Evol. Biol. 2016, 16, 7. [Google Scholar] [CrossRef]
- Rahman, S.; Clarke, C.F.; Hirano, M. 176th ENMC International Workshop: Diagnosis and Treatment of Coenzyme Q10 Deficiency. Neuromuscul. Disord. 2012, 22, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Maawali, A.; Blaser, S.; Yoon, G. Diagnostic Approach to Childhood-Onset Cerebellar Atrophy. J. Child Neurol. 2012, 27, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Q.E.; Tan, T.S.; Kawamukai, M.; Chen, E.S. Cellular Factories for Coenzyme Q10 Production. Microb. Cell Fact. 2017, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; DiMauro, S.; Hirano, M. Treatment of CoQ10 Deficient Fibroblasts with Ubiquinone, CoQ Analogs, and Vitamin C: Time- and Compound-Dependent Effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef] [Green Version]
- Cluis, C.P.; Burja, A.M.; Martin, V.J.J. Current Prospects for the Production of Coenzyme Q10 in Microbes. Trends Biotechnol. 2007, 25, 514–521. [Google Scholar] [CrossRef]
- López-Lluch, G.; Del Pozo-Cruz, J.; Sánchez-Cuesta, A.; Cortés-Rodríguez, A.B.; Navas, P. Bioavailability of Coenzyme Q10 Supplements Depends on Carrier Lipids and Solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef]
- Ikematsu, H.; Nakamura, K.; Harashima, S.; Fujii, K.; Fukutomi, N. Safety Assessment of Coenzyme Q10 (Kaneka Q10) in Healthy Subjects: A Double-Blind, Randomized, Placebo-Controlled Trial. Regul. Toxicol. Pharmacol. 2006, 44, 212–218. [Google Scholar] [CrossRef]
- Shults, C.W.; Beal, M.F.; Fontaine, D.; Nakano, K.; Haas, R.H. Absorption, Tolerability, and Effects on Mitochondrial Activity of Oral Coenzyme Q10 in Parkinsonian Patients. Neurology 1998, 50, 793–795. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of Coenzyme Q10 in Early Parkinson Disease: Evidence of Slowing of the Functional Decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Nashimoto, S.; Takekawa, Y.; Takekuma, Y.; Sugawara, M.; Sato, Y. Transport via Niemann-Pick C1 Like 1 Contributes to the Intestinal Absorption of Ubiquinone. Drug Metab. Pharmacokinet. 2020, 35, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Matson, S.; Matson, W.R.; Cormier, K.; Del Signore, S.J.; Hagerty, S.W.; Stack, E.C.; Ryu, H.; Ferrante, R.J. Dose Ranging and Efficacy Study of High-Dose Coenzyme Q10 Formulations in Huntington’s Disease Mice. Biochim. Biophys. Acta 2006, 1762, 616–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 Administration Increases Brain Mitochondrial Concentrations and Exerts Neuroprotective Effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [Green Version]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Ubiquinol-10 Ameliorates Mitochondrial Encephalopathy Associated with CoQ Deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Luo, K.; Quan, Y.; Lim, S.W.; Shin, Y.J.; Lee, K.E.; Kim, H.L.; Ko, E.J.; Kim, J.H.; Chung, S.J.; et al. Water-Soluble Coenzyme Q10 Provides Better Protection than Lipid-Soluble Coenzyme Q10 in a Rat Model of Chronic Tacrolimus Nephropathy. Korean J. Intern. Med. 2021, 36, 949–961. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Zhang, X.; Yang, S.; Liu, Z.; Zhang, J.; Chen, X. Formulation, Characterization, Pharmacokinetics and Antioxidant Activity Evaluation of Pinus Koraiensis Nuts Oil Based Coenzyme Q10 Loaded Nanoemulsion. Ind. Crops Prod. 2022, 187, 115444. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Y.; Cui, Q.; Cheng, J.; Killpartrik, A.; Kemp, A.H.; Guo, M. Characterization, Antioxidant Capacity, and Bioaccessibility of Coenzyme Q10 Loaded Whey Protein Nanoparticles. LWT 2022, 160, 113258. [Google Scholar] [CrossRef]
- Ehrenhaus Masotta, N.; Höcht, C.; Contin, M.; Lucangioli, S.; Rojas, A.M.; Tripodi, V.P. Bioavailability of Coenzyme Q10 Loaded in an Oleogel Formulation for Oral Therapy: Comparison with a Commercial-Grade Solid Formulation. Int. J. Pharm. 2020, 582, 119315. [Google Scholar] [CrossRef]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ10 Deficient Endothelial Cell Culture Model for the Investigation of CoQ10 Blood–Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Chhitij, T.; Seo, J.-E.; Keum, T.; Noh, G.; Bashyal, S.; Lamichhane, S.; Kim, J.H.; Lee, J.H.; Park, J.H.; Choi, J.; et al. Optimized Self-Microemulsifying Drug Delivery System Improves the Oral Bioavailability and Brain Delivery of Coenzyme Q 10. Drug Deliv. 2022, 29, 2330–2342. [Google Scholar] [CrossRef]
- Sheykhhasan, M.; Amini, R.; Soleimani Asl, S.; Saidijam, M.; Hashemi, S.M.; Najafi, R. Neuroprotective Effects of Coenzyme Q10-Loaded Exosomes Obtained from Adipose-Derived Stem Cells in a Rat Model of Alzheimer’s Disease. Biomed. Pharmacother. 2022, 152, 113224. [Google Scholar] [CrossRef] [PubMed]
- Auré, K.; Benoist, J.F.; Ogier de Baulny, H.; Romero, N.B.; Rigal, O.; Lombès, A. Progression despite Replacement of a Myopathic Form of Coenzyme Q10 Defect. Neurology 2004, 63, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, C.; Casarin, A.; Pierrel, F.; Vazquez Fonseca, L.; Viola, G.; Salviati, L.; Trevisson, E. Vitamin K2 Cannot Substitute Coenzyme Q10 as Electron Carrier in the Mitochondrial Respiratory Chain of Mammalian Cells. Sci. Rep. 2019, 9, 6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, J.; Saldanha, J.W.; Gould, A.P. A Drosophila Model for Primary Coenzyme Q Deficiency and Dietary Rescue in the Developing Nervous System. Dis. Model. Mech. 2010, 3, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Rodenburg, R.J.; Mayatepek, E.; Distelmaier, F. 4-Hydroxybenzoic Acid Restores CoQ10 Biosynthesis in Human COQ2 Deficiency. Ann. Clin. Transl. Neurol. 2017, 4, 902–908. [Google Scholar] [CrossRef]
- Acosta Lopez, M.J.; Trevisson, E.; Canton, M.; Vazquez-Fonseca, L.; Morbidoni, V.; Baschiera, E.; Frasson, C.; Pelosi, L.; Rascalou, B.; Desbats, M.A.; et al. Vanillic Acid Restores Coenzyme Q Biosynthesis and ATP Production in Human Cells Lacking COQ6. Oxid. Med. Cell. Longev. 2019, 2019, 3904905. [Google Scholar] [CrossRef] [Green Version]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Bünning, G.; Freyer, C.; Prokisch, H.; Karall, D.; Wredenberg, A.; Wedell, A.; López, L.C.; et al. Detection of 6-Demethoxyubiquinone in CoQ10 Deficiency Disorders: Insights into Enzyme Interactions and Identification of Potential Therapeutics. Mol. Genet. Metab. 2017, 121, 216–223. [Google Scholar] [CrossRef]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of Primary Ubiquinone Deficiency Due to a Novel COQ7 Defect Using 2,4–Dihydroxybensoic Acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Luna-Sánchez, M.; Díaz-Casado, E.; Barca, E.; Tejada, M.Á.; Montilla-García, Á.; Cobos, E.J.; Escames, G.; Acuña-Castroviejo, D.; Quinzii, C.M.; López, L.C. The Clinical Heterogeneity of Coenzyme Q10 Deficiency Results from Genotypic Differences in the Coq9 Gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Hashemi, S.S.; Zare-Abdollahi, D.; Bakhshandeh, M.K.; Vafaee, A.; Abolhasani, S.; Inanloo Rahatloo, K.; DanaeeFard, F.; Farboodi, N.; Rohani, M.; Alavi, A. Clinical Spectrum in Multiple Families with Primary COQ10 Deficiency. Am. J. Med. Genet. Part A 2021, 185, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-M.; Na, J.-H.; Park, S.; Lee, Y.-M. Clinical Characteristics of Early-Onset and Late-Onset Leigh Syndrome. Front. Neurol. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yoo, T.; Lee, M.; Lee, Y.; Jeon, E.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J. Genetic Heterogeneity in Leigh Syndrome: Highlighting Treatable and Novel Genetic Causes. Clin. Genet. 2020, 97, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.Z.; Ng, Y.S.; Blain, A.; Jiminez-Moreno, C.; Alston, C.L.; Nesbitt, V.; Simmons, L.; Santra, S.; Wassmer, E.; Blakely, E.L.; et al. Natural History of Leigh Syndrome: A Study of Disease Burden and Progression. Ann. Neurol. 2022, 91, 117–130. [Google Scholar] [CrossRef]
- Tauber, J.; Polla, D.J.; Park, S. Exophthalmos in Kearns-Sayre Syndrome. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2019, 23, 295–297. [Google Scholar] [CrossRef]
- Park, S.B.; Ma, K.T.; Kook, K.H.; Lee, S.Y. Kearns-Sayre Syndrome: -3 Case Reports and Review of Clinical Feature. Yonsei Med. J. 2004, 45, 727. [Google Scholar] [CrossRef] [Green Version]
- Brackmann, F.; Abicht, A.; Ahting, U.; Schröder, R.; Trollmann, R. Classical MERRF Phenotype Associated with Mitochondrial TRNALeu (m.3243A>G) Mutation. Eur. J. Pediatr. 2012, 171, 859–862. [Google Scholar] [CrossRef]
- Ozawa, M.; Nishino, I.; Horai, S.; Nonaka, I.; Goto, Y.-I. Myoclonus Epilepsy Associated with Ragged-Red Fibers: A G-to-A Mutation at Nucleotide Pair 8363 in Mitochondrial TRNALys in Two Families. Muscle Nerve 1997, 20, 271–278. [Google Scholar] [CrossRef]
- Ito, S.; Shirai, W.; Asahina, M.; Hattori, T. Clinical and Brain MR Imaging Features Focusing on the Brain Stem and Cerebellum in Patients with Myoclonic Epilepsy with Ragged-Red Fibers Due to Mitochondrial A8344G Mutation. AJNR. Am. J. Neuroradiol. 2008, 29, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Stumpf, J.D.; Saneto, R.P.; Copeland, W.C. Clinical and Molecular Features of POLG-Related Mitochondrial Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a011395. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A New Mitochondrial Disease Associated with Mitochondrial DNA Heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar] [PubMed]
- Lopez-Gallardo, E.; Solano, A.; Herrero-Martin, M.D.; Martinez-Romero, I.; Castano-Perez, M.D.; Andreu, A.L.; Herrera, A.; Lopez-Perez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP Syndrome in a Patient Harbouring an Insertion in the MT-ATP6 Gene That Results in a Truncated Protein. J. Med. Genet. 2008, 46, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Duno, M.; Wibrand, F.; Baggesen, K.; Rosenberg, T.; Kjaer, N.; Frederiksen, A.L. A Novel Mitochondrial Mutation m.8989G>C Associated with Neuropathy, Ataxia, Retinitis Pigmentosa—The NARP Syndrome. Gene 2013, 515, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanisch, F.; Kornhuber, M.; Alston, C.L.; Taylor, R.W.; Deschauer, M.; Zierz, S. SANDO Syndrome in a Cohort of 107 Patients with CPEO and Mitochondrial DNA Deletions. J. Neurol. Neurosurg. Psychiatry 2015, 86, 630–634. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paprocka, J.; Nowak, M.; Chuchra, P.; Śmigiel, R. COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency. Metabolites 2022, 12, 955. https://doi.org/10.3390/metabo12100955
Paprocka J, Nowak M, Chuchra P, Śmigiel R. COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency. Metabolites. 2022; 12(10):955. https://doi.org/10.3390/metabo12100955
Chicago/Turabian StylePaprocka, Justyna, Magdalena Nowak, Piotr Chuchra, and Robert Śmigiel. 2022. "COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency" Metabolites 12, no. 10: 955. https://doi.org/10.3390/metabo12100955
APA StylePaprocka, J., Nowak, M., Chuchra, P., & Śmigiel, R. (2022). COQ8A-Ataxia as a Manifestation of Primary Coenzyme Q Deficiency. Metabolites, 12(10), 955. https://doi.org/10.3390/metabo12100955