
A Comprehensive Evaluation of Metabolomics Data Preprocessing Methods for Deep Learning



Abstract
:1. Introduction
2. Results and Discussion
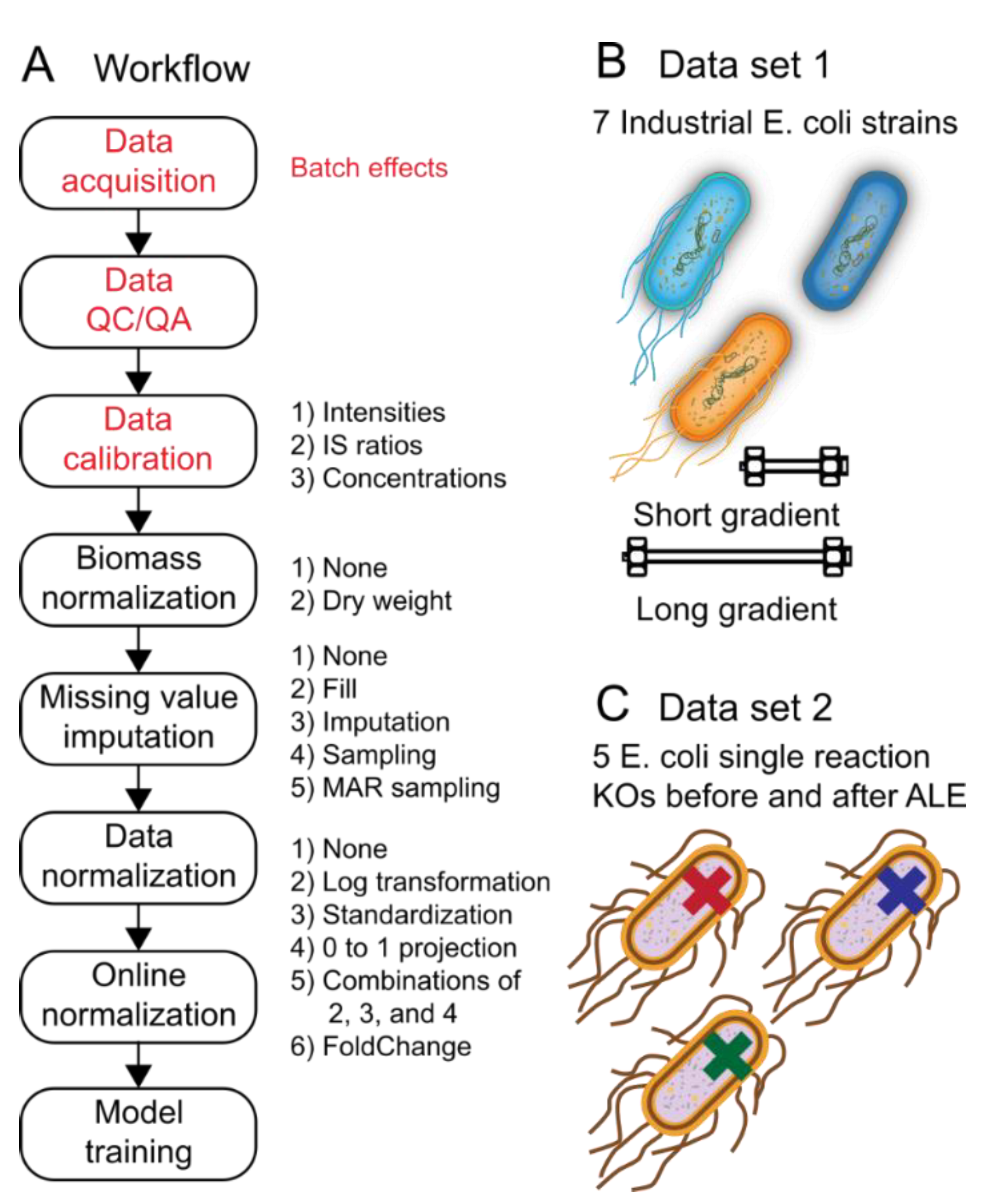
2.1. Overview of the Metabolomics Workflow, Datasets, and Experiments
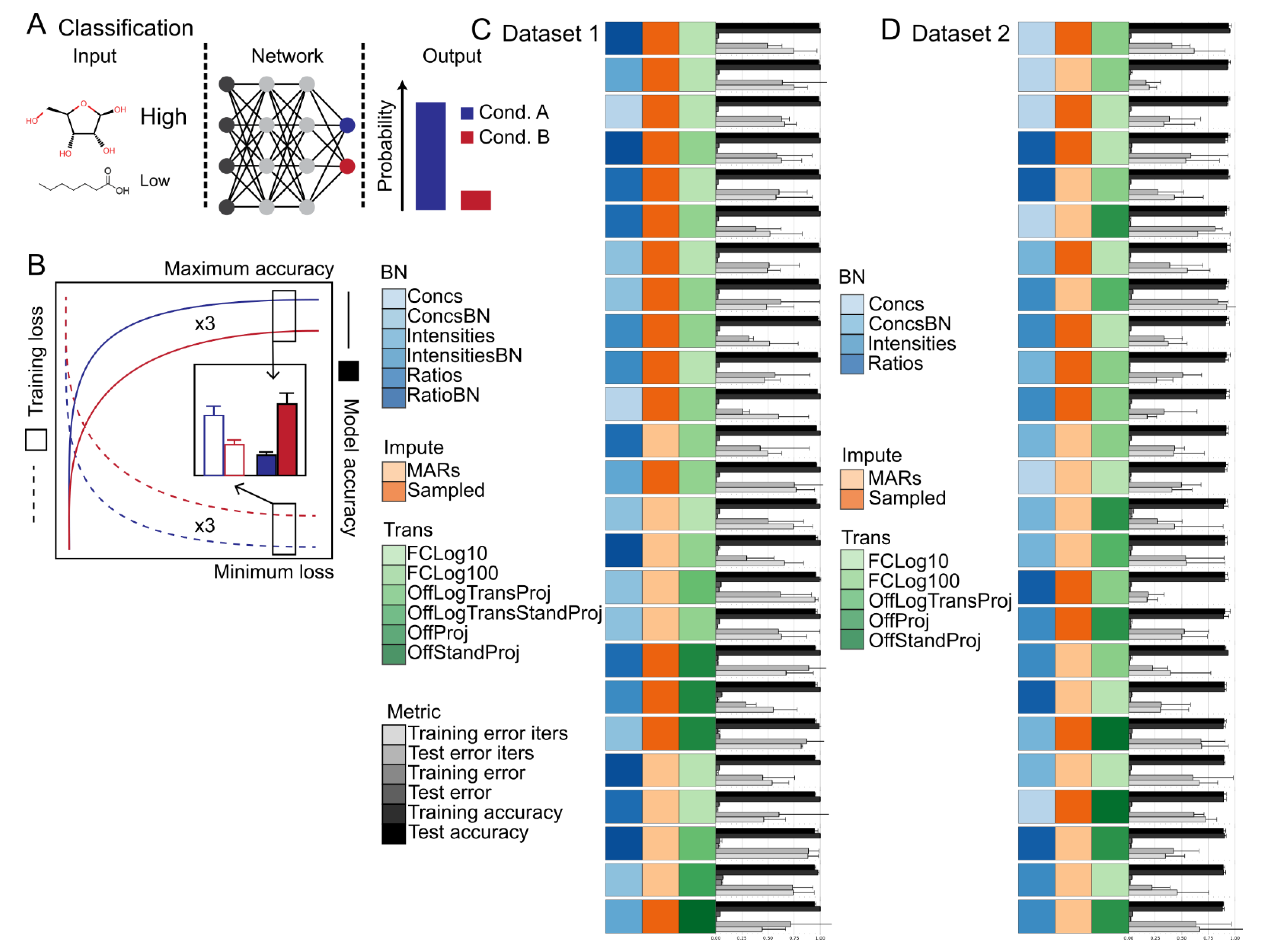
2.2. Data Calibration and Biomass Normalization Have Subtle Influence on Classification Accuracy and Training Speed
- raw ion counts for each metabolite are integrated into peaks and reported as the peak apex or peak area (Intensities),
- the intensities of the metabolite are normalized to the intensities of their corresponding internal standard (Ratios), and
- concentrations are calculated for each metabolite based on an externally ran calibration curve consisting of standards with known amounts (Concs).
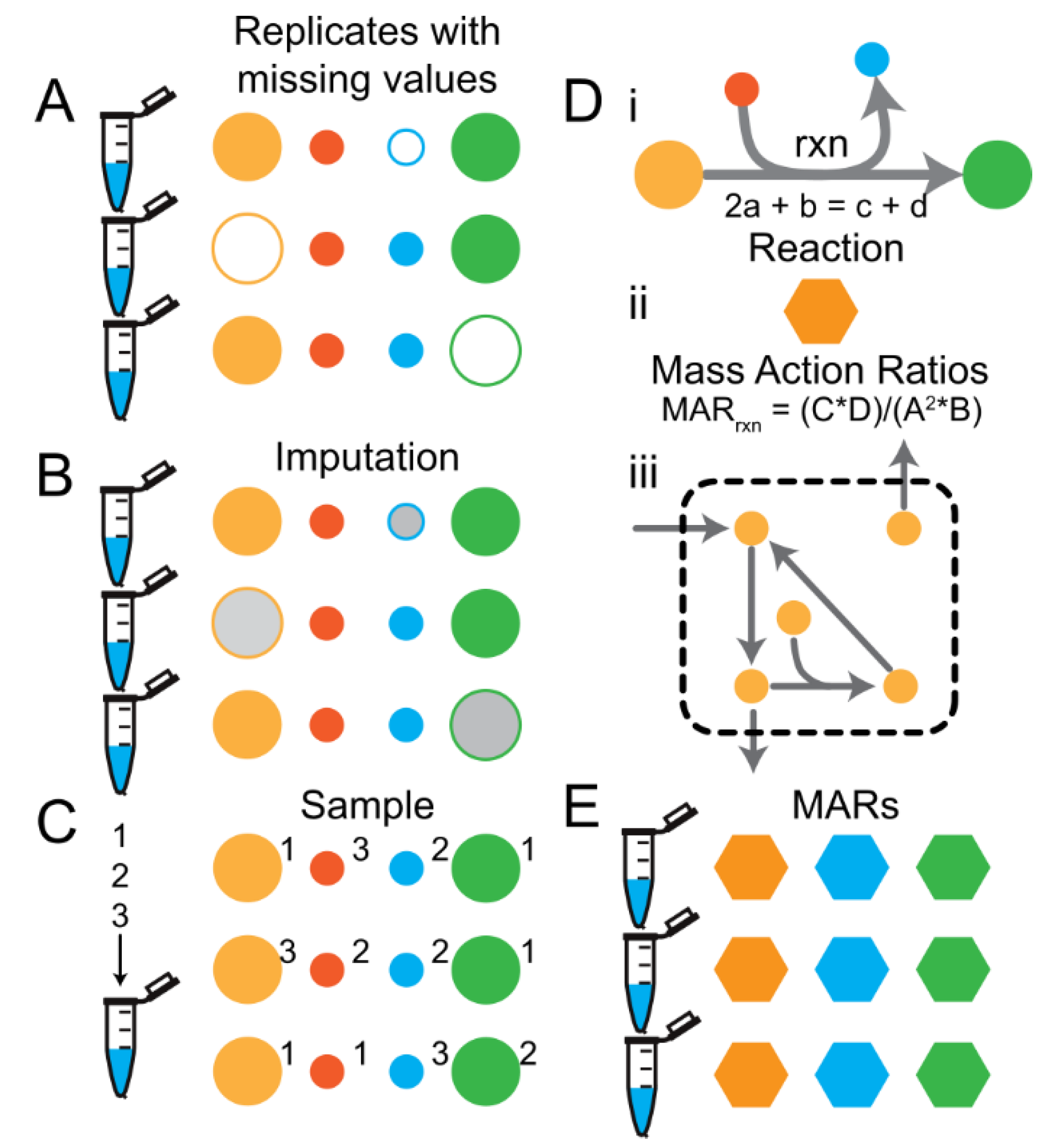
2.3. Sampling-Based Methods Provide a Simple and Powerful Missing Value Imputation Strategy
- Filling with zeros (FillZero),
- Imputing missing values using a probabilistic model (ImputedAmelia, see methods),
- Sampling, and
- Casting the data to mass action ratios (MARs) and sampling the data to compute the MARs (Figure 2D,E).
2.4. Fold-Change Transformation Showed Consistent Superiority to Other Data Transformation Methods
- No normalization (None),
- Projection to the range 0 to 1 (Proj),
- Log normalization followed by projection (LogTransProj),
- Standardization followed by projection (StandProj),
- Log normalization followed by standardization followed by projection (LogTransStandProj),
- Log fold change using bases of 2, 10, or 100 for the base term (FCLogX where X indicates the base used).
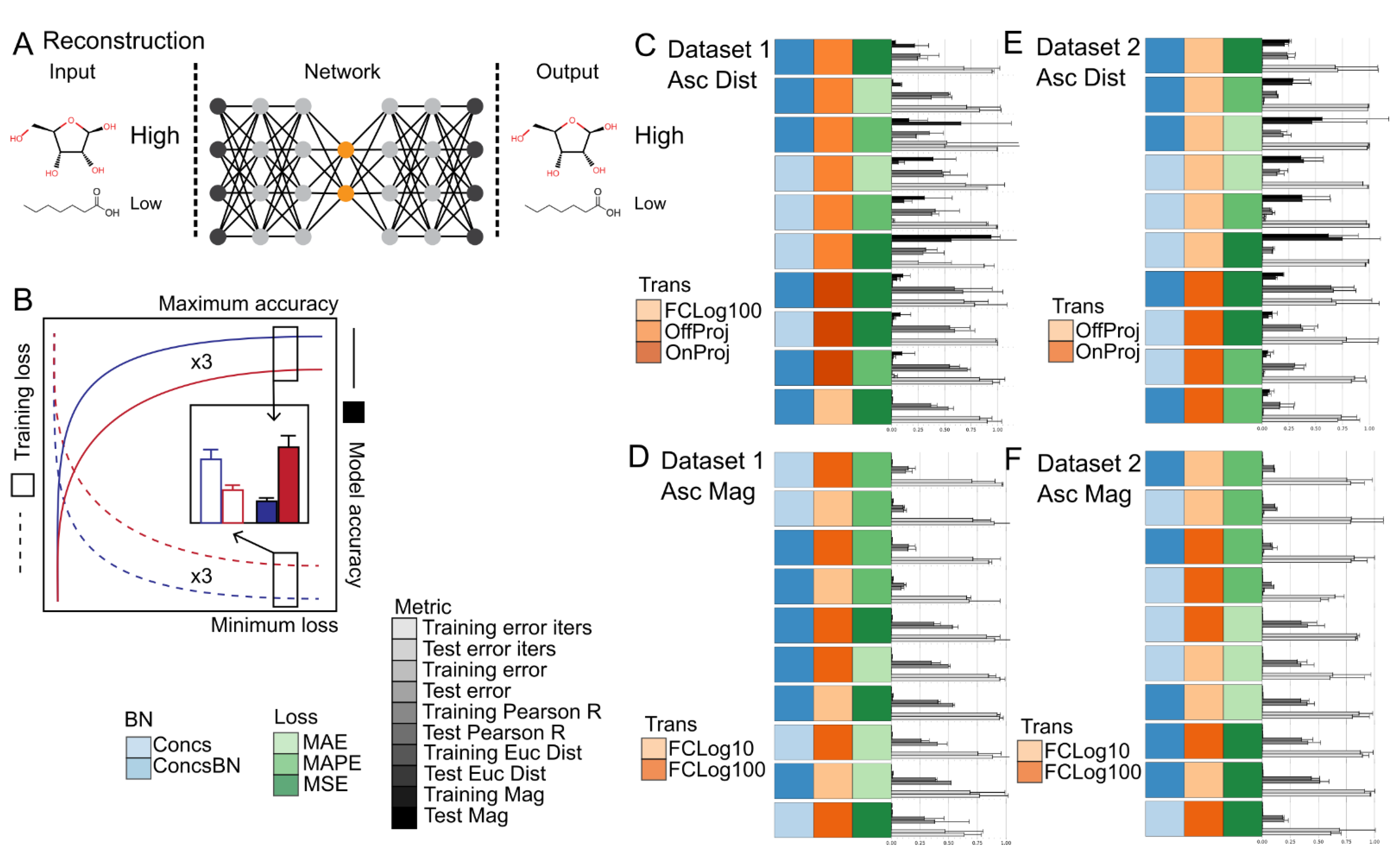
2.5. Reconstruction Accuracy Was Highly Dependent upon the Evaluation Metric Used
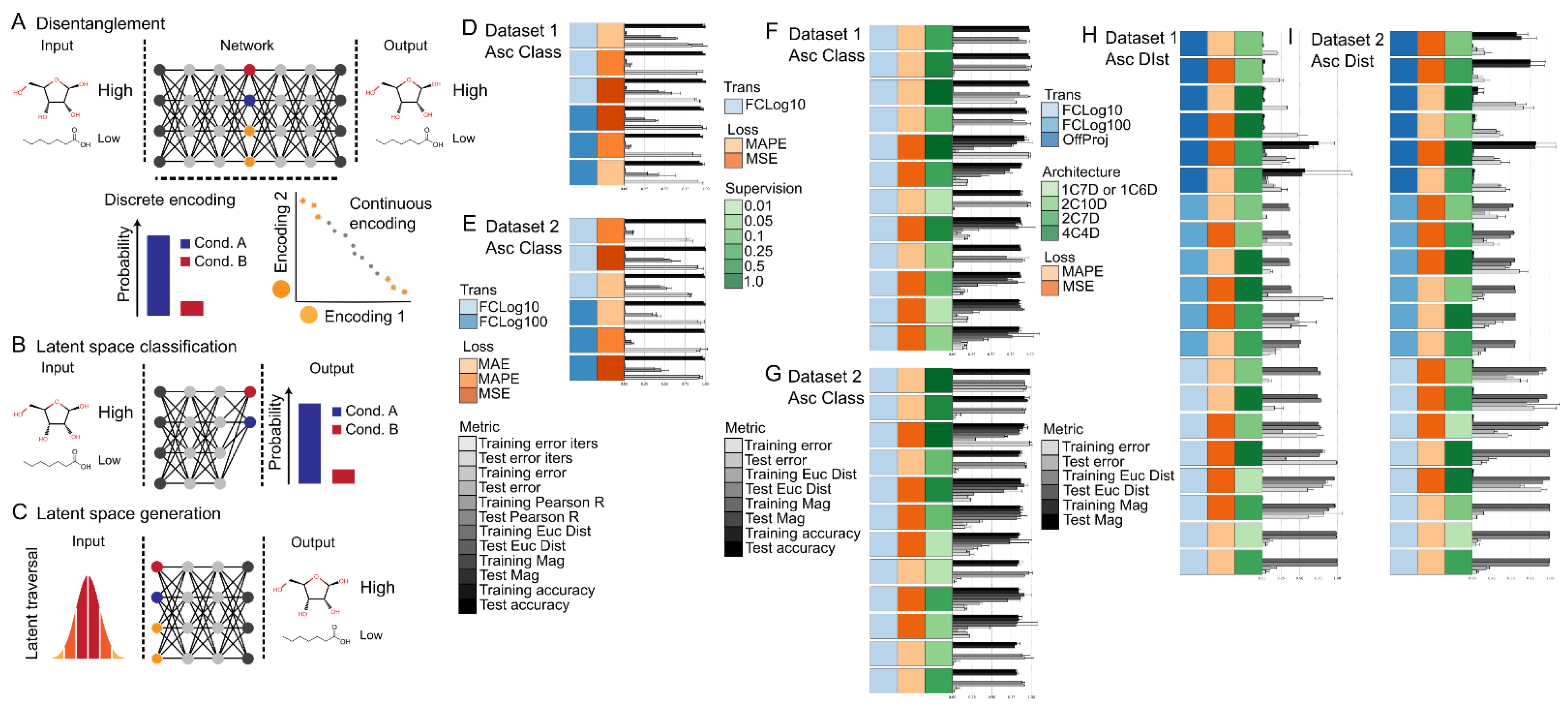
2.6. (Semi-/Un-)Supervised Latent Space Disentanglement of Class and Style Attributes
3. Methods
3.1. Datasets
3.2. Data Preprocessing
3.3. Model and Training Details
3.4. Hyperparameter Tuning
3.5. Code Availability
4. Conclusions
- Differences in biomass can be accounted for using deep learning, but data calibration is needed to account for sample handling and data acquisition effects.
- A sampling strategy to account for missing values provided superior performance to other missing value imputation methods.
- Log fold-change transformation with an optimized base provided superior performance to alternative data normalization methods.
- The optimal combination of data normalization and loss function is dependent upon the reconstruction evaluation metric chosen: Offline project of the dataset to between 0 and 1 to improve numerical stability in combination with MSE or MAE loss was superior for distance-based metrics, while log fold-change transformations in combination with MAPE loss was superior for feature magnitude metrics.
- Log fold-change transformations in combination with MAPE loss provided superior performance for joint reconstruction and classification tasks.
- Unsupervised disentanglement of Omics latent features is a challenging task that requires some level of semi-supervision or prior information to work well.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goh, A.T.C. Back-propagation neural networks for modeling complex systems. Artif. Intell. Eng. 1995, 9, 143–151. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Hestness, J.; Narang, S.; Ardalani, N.; Diamos, G.; Jun, H.; Kianinejad, H.; Patwary, M.M.A.; Yang, Y.; Zhou, Y. Deep Learning Scaling is Predictable, Empirically. arXiv 2017, arXiv:1712.00409. [Google Scholar]
- Shorten, C.; Khoshgoftaar, T.M. A survey on Image Data Augmentation for Deep Learning. J. Big Data 2019, 6, 1–48. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- LeCun, Y.; Bengio, Y. Others Convolutional networks for images, speech, and time series. Handb. Brain Theory Neural Netw. 1995, 3361, 1995. [Google Scholar]
- Silver, D.; Schrittwieser, J.; Simonyan, K.; Antonoglou, I.; Huang, A.; Guez, A.; Hubert, T.; Baker, L.; Lai, M.; Bolton, A.; et al. Mastering the game of Go without human knowledge. Nature 2017, 550, 354–359. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 7873, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 6557, 871–876. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Wolf, F.A.; Theis, F.J. scGen predicts single-cell perturbation responses. Nat. Methods 2019, 16, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Gomari, D.P.; Schweickart, A.; Cerchietti, L.; Paietta, E.; Fernandez, H.; Al-Amin, H.; Suhre, K.; Krumsiek, J. Variational autoencoders learn universal latent representations of metabolomics data. biorXiv 2021. [Google Scholar] [CrossRef]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Burla, B.; Arita, M.; Arita, M.; Bendt, A.K.; Cazenave-Gassiot, A.; Dennis, E.A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G.; et al. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59, 2001–2017. [Google Scholar] [CrossRef] [Green Version]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-based Metabolomics Data. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wang, Y.; Zhang, Y.; Li, F.; Xia, W.; Zhou, Y.; Qiu, Y.; Li, H.; Zhu, F. NOREVA: Enhanced normalization and evaluation of time-course and multi-class metabolomic data. Nucleic Acids Res. 2020, 48, W436–W448. [Google Scholar] [CrossRef]
- Cook, T.; Ma, Y.; Gamagedara, S. Evaluation of statistical techniques to normalize mass spectrometry-based urinary metabolomics data. J. Pharm. Biomed. Anal. 2020, 177, 112854. [Google Scholar] [CrossRef]
- Li, B.; Tang, J.; Yang, Q.; Cui, X.; Li, S.; Chen, S.; Cao, Q.; Xue, W.; Chen, N.; Zhu, F. Performance Evaluation and Online Realization of Data-driven Normalization Methods Used in LC/MS based Untargeted Metabolomics Analysis. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Riekeberg, E.; Qiu, Y.; Powers, R. Comparing normalization methods and the impact of noise. Metabolomics 2018, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Angelov, P.P.; Kangin, D.; Principe, J.C. A new type of distance metric and its use for clustering. Evol. Syst. 2017, 8, 167–177. [Google Scholar] [CrossRef]
- Xing, E.; Jordan, M.; Russell, S.J.; Ng, A. Distance Metric Learning with Application to Clustering with Side-Information. In Proceedings of the Advances in Neural Information Processing Systems; Becker, S., Thrun, S., Obermayer, K., Eds.; MIT Press: Cambridge, MA, USA, 2003; Volume 15. [Google Scholar]
- Dupont, E. Learning Disentangled Joint Continuous and Discrete Representations. arXiv 2018, arXiv:1804.00104. [Google Scholar]
- Tran, D.; Nguyen, H.; Tran, B.; La Vecchia, C.; Luu, H.N.; Nguyen, T. Fast and precise single-cell data analysis using a hierarchical autoencoder. Nat. Commun. 2021, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Fu, X.; Sidiropoulos, N.D.; Hong, M. Towards K-means-friendly Spaces: Simultaneous Deep Learning and Clustering. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; Precup, D., Teh, Y.W., Eds.; PMLR: Cambridge, MA, USA, 2017; Volume 70, pp. 3861–3870. [Google Scholar]
- McCloskey, D.; Xu, J.; Schrübbers, L.; Christensen, H.B.; Herrgård, M.J. RapidRIP quantifies the intracellular metabolome of 7 industrial strains of E. coli. Metab. Eng. 2018, 47, 383–392. [Google Scholar] [CrossRef]
- McCloskey, D.; Xu, S.; Sandberg, T.E.; Brunk, E.; Hefner, Y.; Szubin, R.; Feist, A.M.; Palsson, B.O. Evolution of gene knockout strains of E. coli reveal regulatory architectures governed by metabolism. Nat. Commun. 2018, 9, 3796. [Google Scholar] [CrossRef]
- Honaker, J.; King, G.; Blackwell, M. Amelia II: A Program for Missing Data. J. Stat. Softw. 2011, 45, 1–47. [Google Scholar] [CrossRef]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B.Ø. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol. Syst. Biol. 2011, 7, 535. [Google Scholar] [CrossRef]
- Maas, A.L.; Hannun, A.Y.; Ng, A.Y. Rectifier nonlinearities improve neural network acoustic models. In Proceedings of the 30th International Conference on Machine Learning, Atlanta, GA, USA, 16–21 June 2013; Volume 28. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving Deep into Rectifiers: Surpassing Human-Level Performance on ImageNet Classification. In Proceedings of the 2015 IEEE International Conference on Computer Vision (ICCV), Santiago, Chile, 7–13 December 2015; pp. 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Split | Type | AVE (Euc) | STDDEV (Euc) | AVE (Perc Diff) | STDDEV (Perc Diff) |
|---|---|---|---|---|---|---|
| Dataset 1 (Industrial) | Train | MAPE | 4.917 | 0.493 | 4.750 | 0.722 |
| MSE | 4.250 | 1.090 | 4.500 | 0.500 | ||
| Test | MAPE | 4.417 | 0.954 | 4.500 | 0.866 | |
| MSE | 4.417 | 0.954 | 4.833 | 0.687 | ||
| Dataset 2 (ALEsKOs) | Train | MAPE | 3.000 | 0.000 | 3.750 | 0.595 |
| MSE | 3.750 | 1.010 | 4.000 | 0.707 | ||
| Test | MAPE | 3.250 | 0.433 | 4.167 | 0.986 | |
| MSE | 4.167 | 0.553 | 3.833 | 0.799 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abram, K.J.; McCloskey, D. A Comprehensive Evaluation of Metabolomics Data Preprocessing Methods for Deep Learning. Metabolites 2022, 12, 202. https://doi.org/10.3390/metabo12030202
Abram KJ, McCloskey D. A Comprehensive Evaluation of Metabolomics Data Preprocessing Methods for Deep Learning. Metabolites. 2022; 12(3):202. https://doi.org/10.3390/metabo12030202
Chicago/Turabian StyleAbram, Krzysztof Jan, and Douglas McCloskey. 2022. "A Comprehensive Evaluation of Metabolomics Data Preprocessing Methods for Deep Learning" Metabolites 12, no. 3: 202. https://doi.org/10.3390/metabo12030202
APA StyleAbram, K. J., & McCloskey, D. (2022). A Comprehensive Evaluation of Metabolomics Data Preprocessing Methods for Deep Learning. Metabolites, 12(3), 202. https://doi.org/10.3390/metabo12030202