
TRACES: A Lightweight Browser for Liquid Chromatography–Multiple Reaction Monitoring–Mass Spectrometry Chromatograms



Abstract
:1. Introduction
2. Results and Discussion
2.1. Software Design Concept and Workflow
2.2. Retention Time Alignment
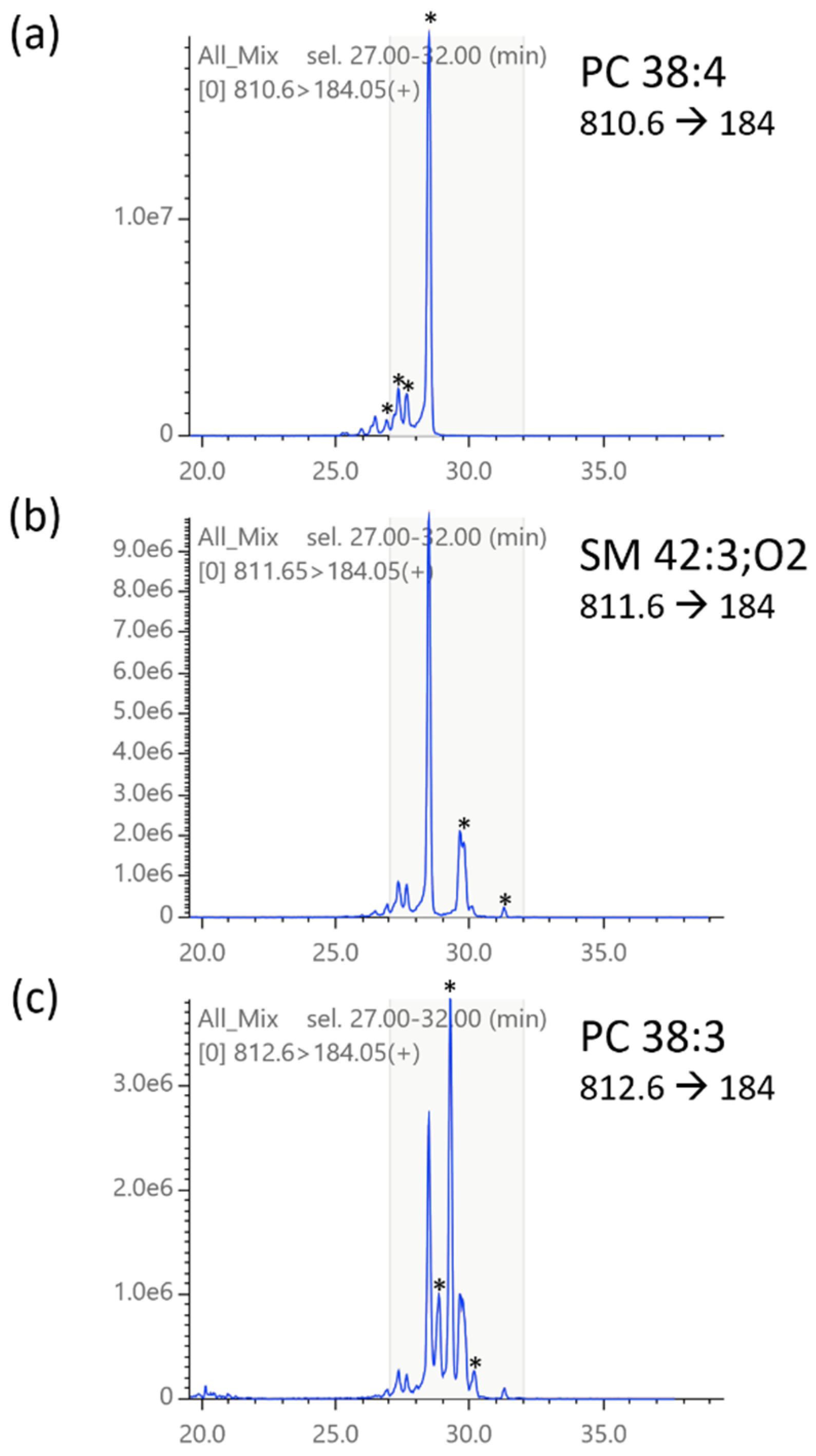
2.3. Compound Library
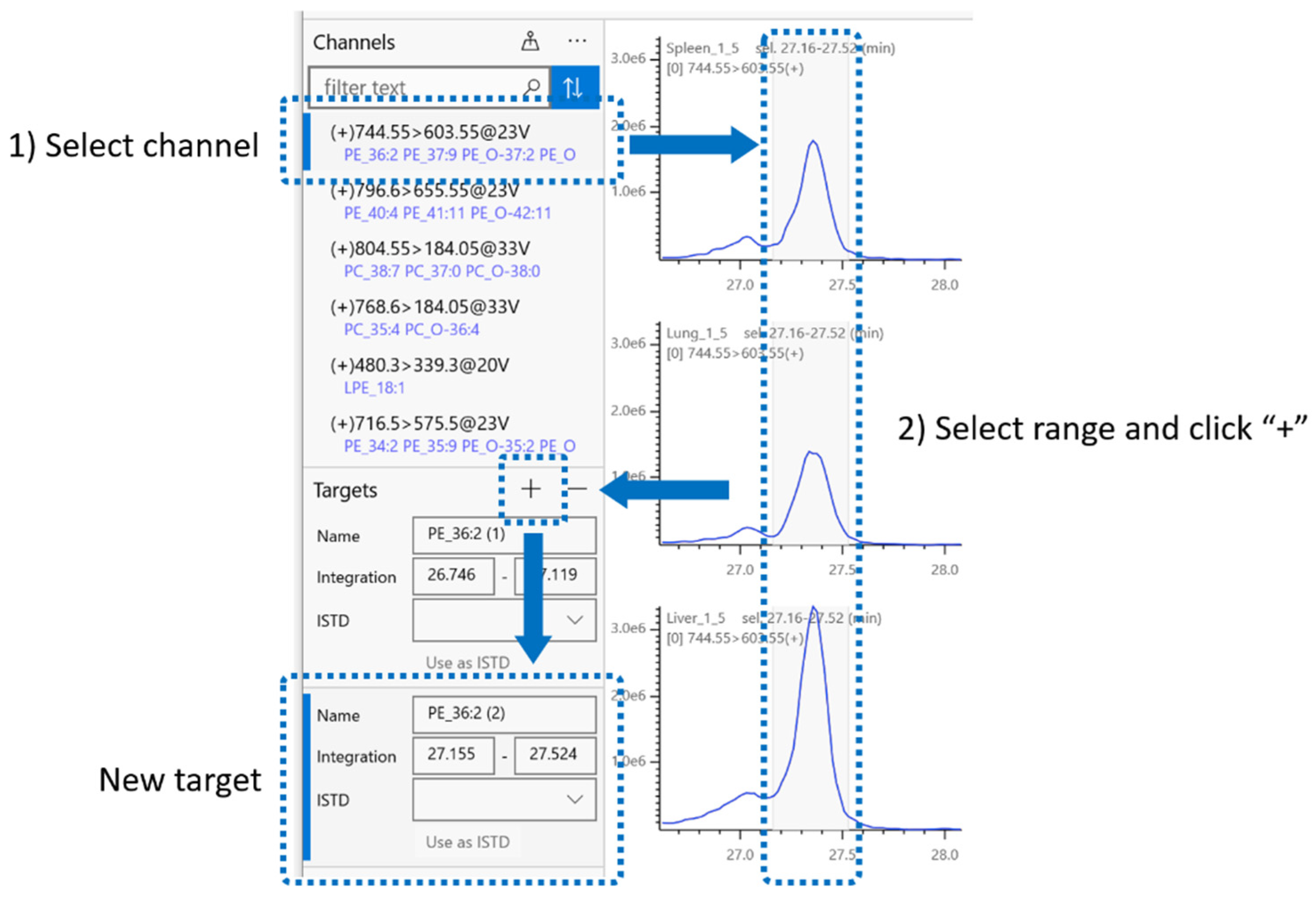
2.4. Integration Target
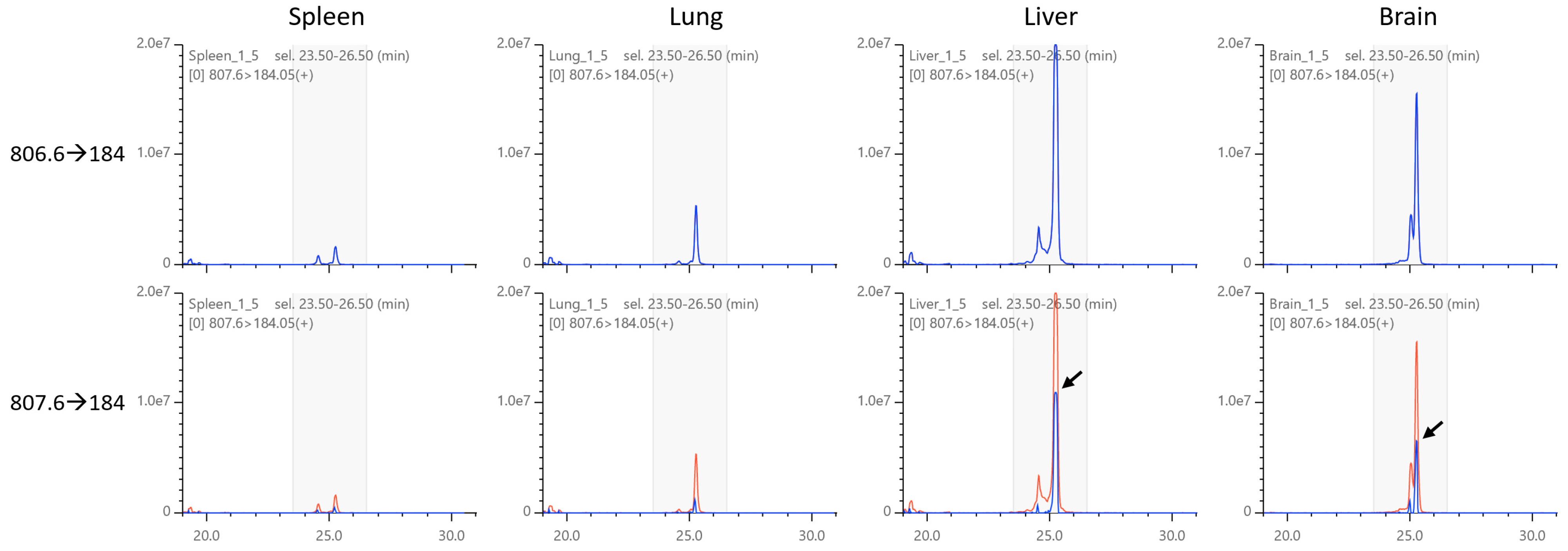
2.5. Isotopic Correction of Chromatograms
2.6. Application to Mouse Phospholipid Analysis
3. Materials and Methods
3.1. Chemicals
3.2. LC-MRM-MS
3.3. Software Implementation
3.4. Theories for MS2-Level Isotopic Distribution and Deisotoping
3.5. Lipid Nomenclature and Notation
3.6. Data Processing and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kita, Y.; Takahashi, T.; Uozumi, N.; Shimizu, T. A Multiplex Quantitation Method for Eicosanoids and Platelet-Activating Factor Using Column-Switching Reversed-Phase Liquid Chromatography–Tandem Mass Spectrometry. Anal. Biochem. 2005, 342, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yin, Y. Strategies for Large-Scale Targeted Metabolomics Quantification by Liquid Chromatography-Mass Spectrometry. Analyst 2016, 141, 6362–6373. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Dai, W.; Yin, P.; Zeng, Z.; Kong, H.; Zhou, L.; Wang, X.; Chen, S.; Lu, X.; Xu, G. Multiple Reaction Monitoring-Ion Pair Finder: A Systematic Approach To Transform Nontargeted Mode to Pseudotargeted Mode for Metabolomics Study Based on Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2015, 87, 5050–5055. [Google Scholar] [CrossRef] [PubMed]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.; Rinner, O.; Picotti, P.; Hüttenhain, R.; Beck, M.; Brusniak, M.-Y.; Hengartner, M.O.; Aebersold, R. MProphet: Automated Data Processing and Statistical Validation for Large-Scale SRM Experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Nasso, S.; Goetze, S.; Martens, L. Ariadne’s Thread: A Robust Software Solution Leading to Automated Absolute and Relative Quantification of SRM Data. J. Proteome Res. 2015, 14, 3779–3792. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Kanazawa, M.; Ogiwara, A.; Arita, M. MRMPROBS Suite for Metabolomics Using Large-Scale MRM Assays. Bioinformatics 2014, 30, 2379–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsugawa, H.; Ohta, E.; Izumi, Y.; Ogiwara, A.; Yukihira, D.; Bamba, T.; Fukusaki, E.; Arita, M. MRM-DIFF: Data Processing Strategy for Differential Analysis in Large Scale MRM-Based Lipidomics Studies. Front. Genet. 2015, 5, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, K.J.; Pratt, B.; Bose, N.; Dubois, L.G.; St. John-Williams, L.; Perrott, K.M.; Ky, K.; Kapahi, P.; Sharma, V.; MacCoss, M.J.; et al. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Chitpin, J.G.; Surendra, A.; Nguyen, T.T.; Taylor, G.P.; Xu, H.; Alecu, I.; Ortega, R.; Tomlinson, J.J.; Crawley, A.M.; McGuinty, M.; et al. BATL: Bayesian Annotations for Targeted Lipidomics. Bioinformatics 2022, 38, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Ji, S.; Burla, B.; Wenk, M.R.; Torta, F.; Cazenave-Gassiot, A. LICAR: An Application for Isotopic Correction of Targeted Lipidomic Data Acquired with Class-Based Chromatographic Separations Using Multiple Reaction Monitoring. Anal. Chem. 2021, 93, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gross, R.W. Shotgun Lipidomics: Electrospray Ionization Mass Spectrometric Analysis and Quantitation of Cellular Lipidomes Directly from Crude Extracts of Biological Samples. Mass Spectrom. Rev. 2005, 24, 367–412. [Google Scholar] [CrossRef]
- Liebisch, G.; Lieser, B.; Rathenberg, J.; Drobnik, W.; Schmitz, G. High-Throughput Quantification of Phosphatidylcholine and Sphingomyelin by Electrospray Ionization Tandem Mass Spectrometry Coupled with Isotope Correction Algorithm. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2004, 1686, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Castellaneta, A.; Losito, I.; Coniglio, D.; Leoni, B.; Santamaria, P.; di Noia, M.A.; Palmieri, L.; Calvano, C.D.; Cataldi, T.R.I. LIPIC: An Automated Workflow to Account for Isotopologue-Related Interferences in Electrospray Ionization High-Resolution Mass Spectra of Phospholipids. J. Am. Soc. Mass Spectrom. 2021, 32, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Hashidate-Yoshida, T.; Harayama, T.; Hishikawa, D.; Morimoto, R.; Hamano, F.; Tokuoka, S.M.; Eto, M.; Tamura-Nakano, M.; Yanobu-Takanashi, R.; Mukumoto, Y.; et al. Fatty Acid Remodeling by LPCAT3 Enriches Arachidonate in Phospholipid Membranes and Regulates Triglyceride Transport. eLife 2015, 4, e06328. [Google Scholar] [CrossRef] [PubMed]
- Valentine, W.J.; Mostafa, S.A.; Tokuoka, S.M.; Hamano, F.; Inagaki, N.F.; Nordin, J.Z.; Motohashi, N.; Kita, Y.; Aoki, Y.; Shimizu, T.; et al. Lipidomic Analyses Reveal Specific Alterations of Phosphatidylcholine in Dystrophic Mdx Muscle. Front. Physiol. 2022, 12, 698166. [Google Scholar] [CrossRef]
- Win2D. Available online: https://github.com/microsoft/Win2D (accessed on 26 March 2022).
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS Classification, Nomenclature, and Shorthand Notation for MS-Derived Lipid Structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- TRACES. Available online: https://github.com/KitaYoshihiro/TRACES/releases/tag/v0.2.48.0-update.1 (accessed on 26 March 2022).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field | Type | Description |
|---|---|---|
| Name | string | Compound name |
| Q1 | numeric | Q1 m/z value |
| Q3 | numeric | Q3 m/z value |
| CE ** | numeric | Collision energy |
| Polarity | string | ‘Positive’ or ‘Negative’ |
| Formula * | string | Formula for precursor ion |
| MS2Formula * | string | Formula for product ion or neutral loss fragment |
| MS2FormulaTyp e * | string | ‘ConstantProduct’ or ‘ConstantNeutralLoss’ |
| Tags ** | string | Any strings for search/filter |
| Compound | Isotopologue | Q1 m/z | MS1 Abundance | Q3 m/z | MS2 Abundance | Affected Compound |
|---|---|---|---|---|---|---|
| PC 34:2 * | M0 | 758.6+ | α | 184+ ** | β | (self) |
| M1 | 759.6+ | 4.7 × 10−1 α | 184+ | 4.1 × 10−1 β | SM 38:1;O2 | |
| 185+ | 6.1 × 10−2 β | - | ||||
| M2 | 760.6+ | 1.2 × 10−1 α | 184+ | 9.0 × 10−2 β | PC 34:1 | |
| 185+ | 2.5 × 10−2 β | - | ||||
| 186+ | 9.8 × 10−3 β | - | ||||
| M3 | 761.6+ | 2.4 × 10−2 α | 184+ | 1.4 × 10−2 β | SM 38:0;O2 | |
| 185+ | 5.4 × 10−3 β | - | ||||
| 186+ | 4.0 × 10−3 β | - | ||||
| 187+ | 5.2 × 10−4 β | - | ||||
| M4 | 762.6+ | 3.7 × 10−3 α | 184+ | 1.7 × 10−3 β | PC 34:0 | |
| 185+ | 8.5 × 10−4 β | - | ||||
| 186+ | 8.8 × 10−4 β | - | ||||
| 187+ | 2.1 × 10−4 β | - | ||||
| 188+ | 3.8 × 10−5 β | - |
| Compound | Isotopologue | Q1 m/z | MS1 Abundance | Q3 m/z | MS2 Abundance | Affected Compound |
|---|---|---|---|---|---|---|
| PS 34:2 * | M0 | 758.5− | α | 673.5− ** | β | (self) |
| M1 | 759.5− | 4.5 × 10−1 α | 673.5− | 3.7 × 10−2 β | - | |
| 674.5− | 4.1 × 10−1 β | - | ||||
| M2 | 760.5− | 1.2 × 10−1 α | 673.5− | 4.6 × 10−3 β | - | |
| 674.5− | 1.5 × 10−2 β | - | ||||
| 675.5− | 9.9 × 10−2 β | PS 34:1 | ||||
| M3 | 761.5− | 2.3 × 10−2 α | 673.5− | 1.6 × 10−4 β | - | |
| 674.5− | 1.9 × 10−3 β | - | ||||
| 675.5− | 3.7 × 10−3 β | - | ||||
| 676.5− | 1.7 × 10−2 β | - | ||||
| M4 | 762.5− | 3.7 × 10−3 α | 673.5− | 6.3 × 10−6 β | - | |
| 674.5− | 6.3 × 10−5 β | - | ||||
| 675.5− | 4.5 × 10−4 β | - | ||||
| 676.5− | 6.5 × 10−4 β | - | ||||
| 677.5− | 2.5 × 10−3 β | PS 34:0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kita, Y.; Tokuoka, S.M.; Oda, Y.; Shimizu, T. TRACES: A Lightweight Browser for Liquid Chromatography–Multiple Reaction Monitoring–Mass Spectrometry Chromatograms. Metabolites 2022, 12, 354. https://doi.org/10.3390/metabo12040354
Kita Y, Tokuoka SM, Oda Y, Shimizu T. TRACES: A Lightweight Browser for Liquid Chromatography–Multiple Reaction Monitoring–Mass Spectrometry Chromatograms. Metabolites. 2022; 12(4):354. https://doi.org/10.3390/metabo12040354
Chicago/Turabian StyleKita, Yoshihiro, Suzumi M. Tokuoka, Yoshiya Oda, and Takao Shimizu. 2022. "TRACES: A Lightweight Browser for Liquid Chromatography–Multiple Reaction Monitoring–Mass Spectrometry Chromatograms" Metabolites 12, no. 4: 354. https://doi.org/10.3390/metabo12040354
APA StyleKita, Y., Tokuoka, S. M., Oda, Y., & Shimizu, T. (2022). TRACES: A Lightweight Browser for Liquid Chromatography–Multiple Reaction Monitoring–Mass Spectrometry Chromatograms. Metabolites, 12(4), 354. https://doi.org/10.3390/metabo12040354