
Folate Related Pathway Gene Analysis Reveals a Novel Metabolic Variant Associated with Alzheimer’s Disease with a Change in Metabolic Profile

 ,
,  ,
,
Abstract
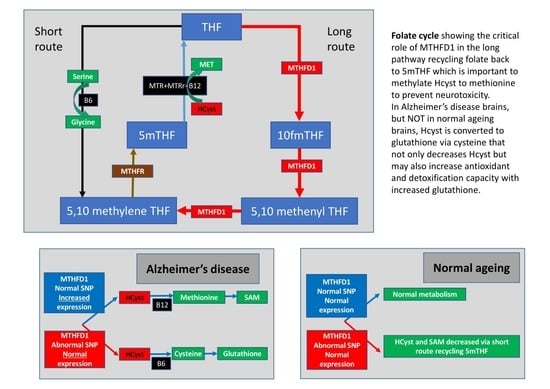
:
1. Introduction
2. Results
2.1. Folate Metabolism
2.2. SNPs in Neural-Related Genes
2.3. SNPs in Folate and Methylation-Related Genes
2.4. Changes in the Metabolic Profile Associated with Folate Gene SNP
3. Discussion
4. Material and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nikolac Perkovic, M. Pivac N: Genetic Markers of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1192, 27–52. [Google Scholar] [PubMed]
- Novikova, G.; Andrews, S.J.; Renton, A.E.; Marcora, E. Beyond association: Successes and challenges in linking non-coding genetic variation to functional consequences that modulate Alzheimer’s disease risk. Mol. Neurodegener 2021, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, A.; Aruoma, O.I. Alzheimer’s Disease and Parkinson's Disease: A Nutritional Toxicology Perspective of the Impact of Oxidative Stress, Mitochondrial Dysfunction, Nutrigenomics and Environmental Chemicals. J. Am. Coll. Nutr. 2020, 39, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Norwitz, N.G.; Saif, N.; Ariza, I.E.; Isaacson, R.S. Precision Nutrition for Alzheimer’s Prevention in ApoE4 Carriers. Nutrients 2021, 13, 1362. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Li, X.; Zhang, S.; Zhao, J.; Zhu, X.; Tian, G. Head Injury as a Risk Factor for Dementia and Alzheimer’s Disease: A Systematic Review and Meta-Analysis of 32 Observational Studies. PLoS ONE 2017, 12, e0169650. [Google Scholar] [CrossRef]
- Nagamine, T. Minor Head Injury Might Cause Treatable Dementia Due to Severe Hyponatremia. Innov. Clin. Neurosci. 2021, 18, 47–48. [Google Scholar]
- Schneider, A.L.C.; Selvin, E.; Latour, L.; Turtzo, L.C.; Coresh, J.; Mosley, T.; Ling, G.; Gottesman, R.F. Head injury and 25-year risk of dementia. Alzheimers Dement 2021, 17, 1432–1441. [Google Scholar] [CrossRef]
- Lemche, E. Early Life Stress and Epigenetics in Late-onset Alzheimer’s Dementia: A Systematic Review. Curr. Genom. 2018, 19, 522–602. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppede, F. Role of epigenetics in Alzheimer’s disease pathogenesis. Neurodegener Dis. Manag. 2018, 8, 181–193. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, X.; Jiao, B. Epigenetics: Recent Advances and Its Role in the Treatment of Alzheimer’s Disease. Front. Neurol. 2020, 11, 538301. [Google Scholar] [CrossRef] [PubMed]
- Cains, S.; Shepherd, A.; Nabiuni, M.; Owen-Lynch, P.J.; Miyan, J. Addressing a folate imbalance in fetal cerebrospinal fluid can decrease the incidence of congenital hydrocephalus. J. Neuropathol. Exp. Neurol. 2009, 68, 404–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requena-Jimenez, A.; Nabiuni, M.; Miyan, J.A. Profound changes in cerebrospinal fluid proteome and metabolic profile are associated with congenital hydrocephalus. J. Cereb. Blood Flow Metab. 2021, 41, 3400–3414. [Google Scholar] [CrossRef] [PubMed]
- Naz, N.; Jimenez, A.R.; Sanjuan-Vilaplana, A.; Gurney, M.; Miyan, J. Neonatal hydrocephalus is a result of a block in folate handling and metabolism involving 10-formyltetrahydrofolate dehydrogenase. J. Neurochem. 2016, 138, 610–623. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.R.; Naz, N.; Miyan, J.A. Altered folate binding protein expression and folate delivery are associated with congenital hydrocephalus in the hydrocephalic Texas rat. J. Cereb. Blood Flow Metab. 2019, 39, 2061–2073. [Google Scholar] [CrossRef] [Green Version]
- Harvey, I.; McGuffin, P.; Williams, M.; Toone, B.K. The ventricle-brain ratio (VBR) in functional psychoses: An admixture analysis. Psychiatry Res. 1990, 35, 61–69. [Google Scholar] [CrossRef]
- Jones, P.B.; Harvey, I.; Lewis, S.W.; Toone, B.K.; Van Os, J.; Williams, M.; Murray, R.M. Cerebral ventricle dimensions as risk factors for schizophrenia and affective psychosis: An epidemiological approach to analysis. Psychol. Med. 1994, 24, 995–1011. [Google Scholar] [CrossRef]
- Saijo, T.; Abe, T.; Someya, Y.; Sassa, T.; Sudo, Y.; Suhara, T.; Shuno, T.; Asai, K.; Okubo, Y. Ten year progressive ventricular enlargement in schizophrenia: An MRI morphometrical study. Psychiatry Clin. Neurosci. 2001, 55, 41–47. [Google Scholar] [CrossRef]
- Strakowski, S.M.; DelBello, M.P.; Zimmerman, M.E.; Getz, G.E.; Mills, N.P.; Ret, J.; Shear, P.; Adler, C.M. Ventricular and periventricular structural volumes in first- versus multiple-episode bipolar disorder. Am. J. Psychiatry 2002, 159, 1841–1847. [Google Scholar] [CrossRef]
- Movsas, T.Z.; Pinto-Martin, J.A.; Whitaker, A.H.; Feldman, J.F.; Lorenz, J.M.; Korzeniewski, S.J.; Levy, S.E.; Paneth, N. Autism spectrum disorder is associated with ventricular enlargement in a low birth weight population. J. Pediatr. 2013, 163, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.D. Cerebrospinal fluid and the early brain development of autism. J. Neurodev. Disord. 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.D.; Nordahl, C.W.; Young, G.S.; Wootton-Gorges, S.L.; Lee, A.; Liston, S.E.; Harrington, K.R.; Ozonoff, S.; Amaral, D.G. Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder. Brain 2013, 136, 2825–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercimek-Mahmutoglu, S.; Stockler-Ipsiroglu, S. Cerebral folate deficiency and folinic acid treatment in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome. Tohoku J. Exp. Med. 2007, 211, 95–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverberg, G.D.; Heit, G.; Huhn, S.; Jaffe, R.A.; Chang, S.D.; Bronte-Stewart, H.; Rubenstein, E.; Possin, K.; Saul, T.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2001, 57, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Ott, B.R.; Cohen, R.A.; Gongvatana, A.; Okonkwo, O.C.; Johanson, C.E.; Stopa, E.G.; Donahue, J.E.; Silverberg, G.D. Alzheimer’s Disease Neuroimaging I: Brain ventricular volume and cerebrospinal fluid biomarkers of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverberg, G.; Mayo, M.; Saul, T.; Fellmann, J.; McGuire, D. Elevated cerebrospinal fluid pressure in patients with Alzheimer’s disease. Cereb. Fluid. Res. 2006, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Nestor, S.M.; Rupsingh, R.; Borrie, M.; Smith, M.; Accomazzi, V.; Wells, J.L.; Fogarty, J.; Bartha, R. Alzheimer’s Disease Neuroimaging I: Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain 2008, 131, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.Y.; Lepore, N.; Avedissian, C.; Madsen, S.K.; Parikshak, N.; Hua, X.; Shaw, L.M.; Trojanowski, J.Q.; Weiner, M.W.; Toga, A.W.; et al. Mapping correlations between ventricular expansion and CSF amyloid and tau biomarkers in 240 subjects with Alzheimer’s disease, mild cognitive impairment and elderly controls. Neuroimage 2009, 46, 394–410. [Google Scholar] [CrossRef] [Green Version]
- Madsen, S.K.; Gutman, B.A.; Joshi, S.H.; Toga, A.W.; Jack, C.R.; Jr Weiner, M.W.; Thompson, P.M. Mapping Dynamic Changes in Ventricular Volume onto Baseline Cortical Surfaces in Normal Aging, MCI, and Alzheimer’s Disease. Multimodal Brain Image Anal. 2013, 8159, 84–94. [Google Scholar]
- Ye, B.S.; Lee, Y.; Kwak, K.; Park, Y.H.; Ham, J.H.; Lee, J.J.; Shin, N.Y.; Lee, J.M.; Sohn, Y.H.; Lee, P.H. Posterior Ventricular Enlargement to Differentiate Dementia with Lewy Bodies from Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 52, 1237–1243. [Google Scholar] [CrossRef]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Iliff, J.J. The impact of neurovascular, blood-brain barrier, and glymphatic dysfunction in neurodegenerative and metabolic diseases. Int. Rev. Neurobiol. 2020, 154, 413–436. [Google Scholar] [PubMed]
- Christensen, J.; Yamakawa, G.R.; Shultz, S.R.; Mychasiuk, R. Is the glymphatic system the missing link between sleep impairments and neurological disorders? Examining the implications and uncertainties. Prog. Neurobiol. 2020, 198, 101917. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef] [PubMed]
- Reeves, B.C.; Karimy, J.K.; Kundishora, A.J.; Mestre, H.; Cerci, H.M.; Matouk, C.; Alper, S.L.; Lundgaard, I.; Nedergaard, M.; Kahle, K.T. Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol. Med. 2020, 26, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Reddy, O.C.; van der Werf, Y.D. The Sleeping Brain: Harnessing the Power of the Glymphatic System through Lifestyle Choices. Brain Sci. 2020, 10, 868. [Google Scholar] [CrossRef]
- Fame, R.M.; Cortes-Campos, C.; Sive, H.L. Brain Ventricular System and Cerebrospinal Fluid Development and Function: Light at the End of the Tube: A Primer with Latest Insights. Bioessays 2020, 42, e1900186. [Google Scholar] [CrossRef] [Green Version]
- Gato, A.; Alonso, M.I.; Lamus, F.; Miyan, J. Neurogenesis: A process ontogenically linked to brain cavities and their content, CSF. Semin. Cell Dev. Biol. 2020, 102, 21–27. [Google Scholar] [CrossRef]
- Miyan, J.; Cains, S.; Larcombe, S.; Naz, N.; Jimenez, A.R.; Bueno, D.; Gato, A. Subarachnoid cerebrospinal fluid is essential for normal development of the cerebral cortex. Semin. Cell Dev. Biol. 2020, 102, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Segers, K.; Sequeira, J.M.; Koenig, M.; Van Maldergem, L.; Bours, V.; Kornak, U.; Quadros, E.V. Genetic assessment and folate receptor autoantibodies in infantile-onset cerebral folate deficiency (CFD) syndrome. Mol. Genet. Metab. 2018, 124, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.; Sequeira, J.M.; Quadros, E.V. Clinical recognition and aspects of the cerebral folate deficiency syndromes. Clin. Chem. Lab. Med. 2013, 51, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Hasselmann, O.; Blau, N.; Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.M.; Weissert, M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol. Genet. Metab. 2010, 99, 58–61. [Google Scholar] [CrossRef]
- Bonkowsky, J.L.; Ramaekers, V.T.; Quadros, E.V.; Lloyd, M. Progressive encephalopathy in a child with cerebral folate deficiency syndrome. J. Child Neurol. 2008, 23, 1460–1463. [Google Scholar] [CrossRef] [Green Version]
- Ramaekers, V.T.; Blau, N.; Sequeira, J.M.; Nassogne, M.C.; Quadros, E.V. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics 2007, 38, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Ramaekers, V.T.; Rothenberg, S.P.; Sequeira, J.M.; Opladen, T.; Blau, N.; Quadros, E.V.; Selhub, J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Engl. J. Med. 2005, 352, 1985–1991. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Delhey, L.; Slattery, J.; Tippett, M.; Wynne, R.; Rose, S.; Kahler, S.G.; Bennuri, S.C.; Melnyk, S.; Sequeira, J.M.; et al. Blocking and Binding Folate Receptor Alpha Autoantibodies Identify Novel Autism Spectrum Disorder Subgroups. Front. Neurosci. 2016, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Sequeira, J.M.; Quadros, E.V.; James, S.J.; Rossignol, D.A. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol. Psychiatry 2013, 18, 369–381. [Google Scholar] [CrossRef]
- Duarte, S.; Cruz Martins, R.; Rodrigues, M.; Lourenco, E.; Moreira, I.; Alonso, I.; Magalhaes, M. Association of cerebral folate deficiency and hereditary spastic paraplegia. Neurologia 2020, 36, 550–552. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.M. Role of folate receptor autoantibodies in infantile autism. Mol. Psychiatry 2013, 18, 270–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafi, S.; Laroche-Raynaud, C.; Chazelas, P.; Lia, A.S.; Derouault, P.; Sturtz, F.; Baaj, Y.; Froget, R.; Rio, M.; Benoist, J.F.; et al. Pharmacoresistant Epilepsy in Childhood: Think of the Cerebral Folate Deficiency, a Treatable Disease. Brain Sci. 2020, 10, 762. [Google Scholar] [CrossRef] [PubMed]
- Leuzzi, V.; Mastrangelo, M.; Celato, A.; Carducci, C.; Carducci, C. A new form of cerebral folate deficiency with severe self-injurious behaviour. Acta Paediatr. 2012, 101, e482–e483. [Google Scholar] [CrossRef]
- Ferreira, P.; Luco, S.M.; Sawyer, S.L.; Davila, J.; Boycott, K.M.; Dyment, D.A. Late diagnosis of cerebral folate deficiency: Fewer seizures with folinic acid in adult siblings. Neurol. Genet. 2016, 2, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadighi, Z.; Butler, I.J.; Koenig, M.K. Adult-onset cerebral folate deficiency. Arch. Neurol. 2012, 69, 778–779. [Google Scholar] [CrossRef] [Green Version]
- Thome, U.; Klima, P.; Moosa, A.N.; Gupta, A.; Parikh, S.; Pestana Knight, E.M. Electrographic status epilepticus in sleep in an adult with cerebral folate deficiency. Neurol. Clin. Pract. 2016, 6, e4–e7. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Wolf, A.; Kim, S.E.; Cabrera, R.M.; Wlodarczyk, B.J.; Zhu, H.; Parker, M.; Lin, Y.; Steele, J.W.; Han, X.; et al. CIC de novo loss of function variants contribute to cerebral folate deficiency by downregulating FOLR1 expression. J. Med. Genet. 2020, 58, 484–494. [Google Scholar] [CrossRef]
- Cario, H.; Smith, D.E.; Blom, H.; Blau, N.; Bode, H.; Holzmann, K.; Pannicke, U.; Hopfner, K.P.; Rump, E.M.; Ayric, Z.; et al. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am. J. Hum. Genet 2011, 88, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Grapp, M.; Just, I.A.; Linnankivi, T.; Wolf, P.; Lucke, T.; Hausler, M.; Gartner, J.; Steinfeld, R. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain 2012, 135, 2022–2031. [Google Scholar] [CrossRef]
- Krsicka, D.; Geryk, J.; Vlckova, M.; Havlovicova, M.; Macek, M., Jr.; Pourova, R. Identification of likely associations between cerebral folate deficiency and complex genetic- and metabolic pathogenesis of autism spectrum disorders by utilization of a pilot interaction modeling approach. Autism Res. 2017, 10, 1424–1435. [Google Scholar] [CrossRef]
- Serrano, M.; Perez-Duenas, B.; Montoya, J.; Ormazabal, A.; Artuch, R. Genetic causes of cerebral folate deficiency: Clinical, biochemical and therapeutic aspects. Drug Discov. Today 2012, 17, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, R.; Grapp, M.; Kraetzner, R.; Dreha-Kulaczewski, S.; Helms, G.; Dechent, P.; Wevers, R.; Grosso, S.; Gartner, J. Folate receptor alpha defect causes cerebral folate transport deficiency: A treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am. J. Hum. Genet 2009, 85, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Luo, J.; Yuan, C.; Ding, D. Vitamin B12, B6, or Folate and Cognitive Function in Community-Dwelling Older Adults: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2020, 77, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Diop-Bove, N.; Visentin, M.; Goldman, I.D. Mechanisms of membrane transport of folates into cells and across epithelia. Annu. Rev. Nutr. 2011, 31, 177–201. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, N.C.; Olsen, S.A.; Dennert, J.W.; Smith, M.R. Ventricular enlargement in schizophrenia: Relationship to positive and negative symptoms. Am. J. Psychiatry 1982, 139, 297–302. [Google Scholar]
- Jayaswal, S.K.; Chawla, H.M.; Goulatia, R.K.; Rao, G.S. Cerebral ventricular enlargement in chronic Schizophrenia. Br. J. Psychiatry 1988, 153, 414–415. [Google Scholar] [CrossRef] [Green Version]
- Chance, S.A.; Esiri, M.M.; Crow, T.J. Ventricular enlargement in schizophrenia: A primary change in the temporal lobe? Schizophr. Res. 2003, 62, 123–131. [Google Scholar] [CrossRef]
- Horga, G.; Bernacer, J.; Dusi, N.; Entis, J.; Chu, K.; Hazlett, E.A.; Haznedar, M.M.; Kemether, E.; Byne, W.; Buchsbaum, M.S. Correlations between ventricular enlargement and gray and white matter volumes of cortex, thalamus, striatum, and internal capsule in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2011, 261, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.; Michelson, D.; Aaen, G.; Ashwal, S. Cerebral folate deficiency presenting as adolescent catatonic schizophrenia: A case report. J. Child Neurol. 2010, 25, 898–900. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Thony, B.; Sequeira, J.M.; Ansseau, M.; Philippe, P.; Boemer, F.; Bours, V.; Quadros, E.V. Folinic acid treatment for schizophrenia associated with folate receptor autoantibodies. Mol. Genet Metab. 2014, 113, 307–314. [Google Scholar] [CrossRef]
- Brennan, L.; de Roos, B. Nutrigenomics: Lessons learned and future perspectives. Am. J. Clin. Nutr. 2021, 113, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Eells, J.T.; Gonzalez-Quevedo, A.; Santiesteban Freixas, R.; McMartin, K.E.; Sadun, A.A. Folic acid deficiency and increased concentrations of formate in serum and cerebrospinal fluid of patients with epidemic optical neuropathy. Rev. Cubana Med. Trop 2000, 52, 21–23. [Google Scholar] [PubMed]
- Anguera, M.C.; Field, M.S.; Perry, C.; Ghandour, H.; Chiang, E.P.; Selhub, J.; Shane, B.; Stover, P.J. Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 2006, 281, 18335–18342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupenko, S.A.; Oleinik, N.V. 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 2002, 13, 227–236. [Google Scholar] [PubMed]
- Smith, D.E.; Hornstra, J.M.; Kok, R.M.; Blom, H.J.; Smulders, Y.M. Folic acid supplementation does not reduce intracellular homocysteine, and may disturb intracellular one-carbon metabolism. Clin. Chem. Lab. Med. 2013, 51, 1643–1650. [Google Scholar] [CrossRef]
- Smith, D.; Hornstra, J.; Rocha, M.; Jansen, G.; Assaraf, Y.; Lasry, I.; Blom, H.; Smulders, Y.M. Folic Acid Impairs the Uptake of 5-Methyltetrahydrofolate in Human Umbilical Vascular Endothelial Cells. J. Cardiovasc. Pharmacol. 2017, 70, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Cornet, D.; Clement, A.; Clement, P.; Menezo, Y. High doses of folic acid induce a pseudo-methylenetetrahydrofolate syndrome. SAGE Open Med. Case Rep. 2019, 7, 2050313X19850435. [Google Scholar] [CrossRef]
- Maruvada, P.; Stover, P.J.; Mason, J.B.; Bailey, R.L.; Davis, C.D.; Field, M.S.; Finnell, R.H.; Garza, C.; Green, R.; Gueant, J.L.; et al. Knowledge gaps in understanding the metabolic and clinical effects of excess folates/folic acid: A summary, and perspectives, from an NIH workshop. Am. J. Clin. Nutr. 2020, 112, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Wharton, S.B.; Wang, D.; Parikh, C.; Matthews, F.E.; Brayne, C.; Ince, P.G.; Cognitive, F. Ageing Neuropathology Study G: Epidemiological pathology of Abeta deposition in the ageing brain in CFAS: Addition of multiple Abeta-derived measures does not improve dementia assessment using logistic regression and machine learning approaches. Acta Neuropathol. Commun. 2019, 7, 198. [Google Scholar] [CrossRef] [Green Version]
- Hunsberger, H.C.; Pinky, P.D.; Smith, W.; Suppiramaniam, V.; Reed, M.N. The role of APOE4 in Alzheimer’s disease: Strategies for future therapeutic interventions. Neuronal. Signal 2019, 3, NS20180203. [Google Scholar] [CrossRef] [Green Version]
- Dorszewska, J.; Florczak, J.; Rozycka, A.; Kempisty, B.; Jaroszewska-Kolecka, J.; Chojnacka, K.; Trzeciak, W.H.; Kozubski, W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson's diseases. Acta Neurobiol. Exp. Wars 2007, 67, 113–129. [Google Scholar] [PubMed]
- Bi, X.H.; Zhao, H.L.; Zhang, Z.X.; Liu, Q.; Zhang, J.W. Association analysis of CbetaS 844ins68 and MTHFD1 G1958A polymorphisms with Alzheimer’s disease in Chinese. J. Neural. Transm. 2010, 117, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xiao, X.; Wen, Y.; Wan, M.; Zhou, L.; Liu, X.; Wang, X.; Guo, L.; Liu, H.; Zhou, Y.; et al. Genetic effect of MTHFR C677T, A1298C, and A1793G polymorphisms on the age at onset, plasma homocysteine, and white matter lesions in Alzheimer’s disease in the Chinese population. Aging 2021, 13, 11352–11362. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, Y.; Liu, X.; Zhou, J.; Wang, Z.; He, Z.; Huang, Z. Lack of association between MTHFR A1298C variant and Alzheimer’s disease: Evidence from a systematic review and cumulative meta-analysis. Neurol. Res. 2017, 39, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Roman, G.C. MTHFR Gene Mutations: A Potential Marker of Late-Onset Alzheimer’s Disease? J. Alzheimer’s Dis. 2015, 47, 323–327. [Google Scholar] [CrossRef]
- Peng, Q.; Lao, X.; Huang, X.; Qin, X.; Li, S.; Zeng, Z. The MTHFR C677T polymorphism contributes to increased risk of Alzheimer’s disease: Evidence based on 40 case-control studies. Neurosci. Lett. 2015, 586, 36–42. [Google Scholar] [CrossRef]
- Pjetri, E.; Zeisel, S.H. Deletion of one allele of Mthfd1 (methylenetetrahydrofolate dehydrogenase 1) impairs learning in mice. Behav. Brain Res. 2017, 332, 71–74. [Google Scholar] [CrossRef]
- Karin, I.; Borggraefe, I.; Catarino, C.B.; Kuhm, C.; Hoertnagel, K.; Biskup, S.; Opladen, T.; Blau, N.; Heinen, F.; Klopstock, T. Folinic acid therapy in cerebral folate deficiency: Marked improvement in an adult patient. J. Neurol. 2017, 264, 578–582. [Google Scholar] [CrossRef]
- Hinterberger, M.; Fischer, P. Folate and Alzheimer: When time matters. J. Neural. Transm. Vienna 2013, 120, 211–224. [Google Scholar] [CrossRef]
- Babic Leko, M.; Nikolac Perkovic, M.; Klepac, N.; Svob Strac, D.; Borovecki, F.; Pivac, N.; Hof, P.R.; Simic, G. Relationships of Cerebrospinal Fluid Alzheimer’s Disease Biomarkers and COMT, DBH, and MAOB Single Nucleotide Polymorphisms. J. Alzheimers Dis. 2020, 73, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.N.; Georrge, J.J.; Modi, K.M.; Narechania, M.B.; Patel, D.P.; Gonzalez, F.J.; Pandya, H.A. Pharmacophore-based virtual screening of catechol-o-methyltransferase (COMT) inhibitors to combat Alzheimer’s disease. J. Biomol. Struct. Dyn. 2018, 36, 3938–3957. [Google Scholar] [CrossRef] [PubMed]
- Zalsman, G.; Huang, Y.Y.; Harkavy-Friedman, J.M.; Oquendo, M.A.; Ellis, S.P.; Mann, J.J. Relationship of MAO-A promoter (u-VNTR) and COMT (V158M) gene polymorphisms to CSF monoamine metabolites levels in a psychiatric sample of caucasians: A preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 132B, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Zalsman, G.; Huang, Y.Y.; Oquendo, M.A.; Brent, D.A.; Giner, L.; Haghighi, F.; Burke, A.K.; Ellis, S.P.; Currier, D.; Mann, J.J. No association of COMT Val158Met polymorphism with suicidal behavior or CSF monoamine metabolites in mood disorders. Arch. Suicide Res. 2008, 12, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Ageing Cases | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Case No. | MRC ID | Gender | Age at Death | Braak Stage | PMD (h) | Clinical Diagnosis | Pathological Diagnosis 1 | Pathological Diagnosis 2 | APOE |
| DPM12/11 | BBN_3478 | M | 54 | 0 | 37 | control | normal brain | 33 | |
| DPM14/04 | BBN_19634 | F | 87 | 0-I | 24 | Normal | Age changes only | 34 | |
| DPM14/08 | BBN_20005 | M | 85 | 0-I | 98 | Normal | Age changes only | moderate SVD | 33 |
| DPM14/20 | BBN_21003 | F | 90 | 0-I | 39 | normal | Age changes only | 33 | |
| DPM14/46 | BBN_24316 | F | 94 | 0-I | 111 | control | age changes only | mild SVD | 23 |
| DPM16/29 | BBN005.29063 | M | 69 | 0-I | 53 | Control | Normal for age | 24 | |
| DPM18/03 | BBN005.32560 | M | 88 | 0-I | 39 | Control | Normal for age | 33 | |
| DPM14/09 | BBN_20006 | M | 84 | I | 69.5 | Normal | Age changes only | moderate SVD | 33 |
| DPM17/09 | BBN005.30100 | F | 88 | I | 52.5 | Control | Normal for age | ARTAG, possible PART | 23 |
| DPM17/34 | BBN005.31485 | M | 89 | I | 125 | Control | Normal for age | Incidental Lewy bodies? | 23 |
| DPM09/31 | BBN_3396 | F | 94 | I-II | cognitively normal/stroke | Age changes only | mild to moderate SVD | 33 | |
| DPM12/09 | BBN_3476 | F | 87 | I-II | 87 | cognitively normal | mild AD pathology in temporal lobe | 33 | |
| DPM13/35 | BBN_15591 | F | 76 | I-II | 47 | normal | mild AD changes in temporal lobe | very mild CAA, moderate SVD in BG | 33 |
| DPM14/11 | BBN_20195 | M | 91 | I-II | 43.5 | Normal | mild SVD | 33 | |
| DPM15/01 | BBN_24368 | M | 90 | I-II | 156 | control | Age changes only | 33 | |
| DPM16/11 | BBN005.28403 | M | 77 | I-II | 63 | Control | Mild temporal tau, possible PART | 33 | |
| DPM16/31 | BBN005.29168 | M | 90 | I-II | 155 | Control | Normal for age | Mild SVD | 33 |
| DPM17/15 | BBN005.30170 | M | 90 | I-II | 125 | Control | Normal for age | Incidental Lewy bodies? | 33 |
| DPM17/36 | BBN005.32382 | F | 94 | I-II | 70 | Control | Age changes only | 33 | |
| DPM18/11 | BBN005.32822 | F | 90 | I-II | 143 | Control | Age changes only | Possible ARTAG | 33 |
| DPM11/06 | BBN_3446 | F | 92 | II | 37 | cognitively normal | Age changes only | mild SVD | 34 |
| DPM11/25 | BBN_3463 | M | 89 | II | 27 | Control | Age changes only | 33 | |
| DPM11/29 | BBN_3467 | M | 89 | II | 123 | cognitively normal | Age changes only | mild SVD | 33 |
| DPM15/28 | BBN_25917 | F | 91 | II | 133 | Control | Age changes only | Cerebral infarction | 23 |
| Severe Alzheimer’s disease cases | |||||||||
| Case No. | MRC ID | Gender | Age at death | Braak stage | PMD (h) | Clinical diagnosis | Pathological diagnosis 1 | Pathological diagnosis 2 | APOE |
| DPM16/16 | BBN005.28547 | F | 81 | V | 176 | AD | AD (Braak V) | V.severe CAA | 34 |
| DPM12/01 | BBN_3469 | M | 67 | V-VI | 84 | Dementia | Alzheimer’s disease | mild SVD | 34 |
| DPM12/32 | BBN_9480 | M | 73 | V-VI | 36 | Alzheimer’s disease | Alzheimer’s disease | 33 | |
| DPM13/09 | BBN_11027 | F | 85 | V-VI | 73 | Alzheimer’s disease | Alzheimer’s disease | moderate to severe SVD, v. Mild DLB | 34 |
| DPM13/10 | BBN_11028 | F | 85 | V-VI | 24 | dementia | Alzheimer’s disease | Mild CAA | 34 |
| DPM13/45 | BBN_19208 | M | 78 | V-VI | 138 | Alzheimer’s disease | Alzheimer’s disease | 33 | |
| DPM14/10 | BBN_20007 | F | 78 | V-VI | 70 | Alzheimer’s disease | Alzheimer’s disease | CAA with capillary involvement | 44 |
| DPM14/50 | BBN_24361 | F | 63 | V-VI | 54 | Alzheimer’s Disease | Alzheimer’s disease | moderate SVD | 44 |
| DPM15/02 | BBN_24373 | M | 78 | V-VI | 173 | Alzheimer’s Disease | Alzheimer’s disease | sec TDP-43 proteinopathy, incidental Lewy bodies? | 44 |
| DPM17/37 | BBN005.32384 | F | 90 | V-VI | 76 | AD | Alzheimer’s disease | Possible AGD | 34 |
| DPM11/28 | BBN_3466 | F | 71 | VI | 64 | Alzheimer’s disease | severe Alzheimer’s disease | 44 | |
| DPM12/03 | BBN_3470 | M | 72 | VI | 81 | Alzheimer’s Disease | Alzheimer’s disease | 34 | |
| DPM12/05 | BBN_3472 | M | 73 | VI | 107 | Alzheimer’s Disease | Alzheimer’s disease | mod SVD | 44 |
| DPM14/30 | BBN_23794 | F | 70 | VI | 89 | dementia, learning difficulty | Alzheimer’s disease | 44 | |
| DPM14/31 | BBN_23803 | M | 64 | VI | 98.5 | Alzheimer’s Disease | Alzheimer’s disease | moderate SVD | 34 |
| DPM15/48 | BBN005.26301 | F | 81 | VI | 98 | Dementia | AD (Braak VI) | Secondary TDP-43 | 34 |
| DPM16/10 | BBN005.28400 | F | 59 | VI | 87 | AD | Alzheimer’s disease | 24 | |
| DPM16/40 | BBN005.29461 | M | 82 | VI | 25.5 | AD | Alzheimer’s disease | Moderate CAA | 34 |
| DPM18/12 | BBN005.32823 | M | 70 | VI | 120.5 | AD? | Alzheimer’s disease | Moderate SVD | 33 |
| DPM18/39 | BBN005.35131 | F | 75 | VI | 127.5 | Dementia | Alzheimer’s disease | 34 | |
| Case No. | MRC ID | Gender | Age at death | Braak stage | PMD (h) | Clinical diagnosis | Pathological diagnosis 1 | Pathological diagnosis 2 | APOE |
| DPM16/16 | BBN005.28547 | F | 81 | V | 176 | AD | AD (Braak V) | V.severe CAA | 34 |
| DPM12/01 | BBN_3469 | M | 67 | V-VI | 84 | Dementia | Alzheimer’s disease | mild SVD | 34 |
| DPM12/32 | BBN_9480 | M | 73 | V-VI | 36 | Alzheimer’s disease | Alzheimer’s disease | 33 | |
| Variable | Control | AD Patients | Total | p |
|---|---|---|---|---|
| Gender | ||||
| Male | 14 | 12 | 26 | |
| Female | 11 | 13 | 24 | 0.571 |
| Sum | 25 | 25 | 50 | |
| Age at death (mean + SD) | 86.5 (9.1) | 75.2 (9.0) | 80.9 (10.6) | 0.0001 |
| Pathological diagnosis 1 | ||||
| Age changes only | 22 | 0 | 22 | |
| Mild temporal lobe pathology | 3 | 0 | 3 | |
| AD typical | 0 | 17 | 17 | |
| AD and dementia | 0 | 6 | 6 | |
| AD and learning difficulty | 0 | 1 | 1 | |
| AD atypical | 0 | 1 | 1 | |
| Pathological diagnosis 2 | ||||
| Mild SVD | 4 | 1 | 5 | |
| Moderate SVD | 4 | 4 | 8 | |
| Severe SVD | 1 | 1 | 2 | |
| Cerebral infarction | 1 | 0 | 1 | |
| Incidental Lewy bodies | 2 | 1 | 3 | |
| Possible ARTAG | 2 | 1 | 3 | |
| CAA | 0 | 4 | 4 | |
| Secondary TDP-43 | 0 | 5 | 5 | |
| Thal phase (n = 29) | ||||
| 0–1 | 9 | 0 | 9 | |
| 2–3 | 5 | 3 | 8 | |
| 4–5 | 0 | 12 | 12 | 0.0003 |
| CERAD score (n = 31) | ||||
| 0 | 6 | 0 | 6 | |
| A | 7 | 0 | 7 | |
| C | 0 | 18 | 18 | <0.0001 |
| Braak stage | ||||
| 0–I | 10 | 0 | 10 | |
| I–II | 15 | 0 | 15 | |
| V–VI | 0 | 25 | 25 | <0.0001 |
| APOE genotype | ||||
| 2/3 | 4 | 0 | 4 | |
| 2/4 | 1 | 1 | 2 | |
| 3/3 | 18 | 6 | 24 | |
| 3/4 | 2 | 11 | 13 | |
| 4/4 | 0 | 7 | 7 | 0.0001 |
| Brain weight, gm (mean ± SD) | 1291.9 (184) | 1133.5 (130) | 1211.0 (176) | 0.0014 |
| PMD (mean ± SD) | 88.8 (45.6) | 100.9 (56.4) | 92.5 (51.6) | 0.2478 |
| Brain pH (mean ± SD) | 5.9 (0.3) | 6.2 (0.5) | 6.1 (0.4) | 0.0617 |
| SNPs | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| OR | 95% CI | p-Value | OR | 95% CI | p-Value | |
| Nervous system related | ||||||
| COMT (rs4633) | 3.80 | 1.418–10.19 | 0.008 | 5.50 | 1.412–21.39 | 0.014 |
| COMT (rs4680) | 3.41 | 1.256–9.251 | 0.016 | 1.00 | (omitted) | |
| CYP2D6 (rs16947) | 2.44 | 1.077–5.544 | 0.033 | 3.64 | 1.135–11.69 | 0.030 |
| ADRB1 (rs1801253) | 2.82 | 0.976–8.133 | 0.055 | 9.61 | 1.781–51.87 | 0.009 |
| _cons | 0.00 | 0.000–0.027 | 0.002 | |||
| Methylation related | ||||||
| MTHFD1 (rs1076991) | 3.98 | 1.550–10.23 | 0.004 | 67.38 | 3.231–1404 | 0.007 |
| MTHFD1 (rs2236225) | 1.52 | 0.726–3.164 | 0.268 | 10.50 | 1.595–69.10 | 0.014 |
| MAT1A (rs1985908) | 0.62 | 0.263–1.448 | 0.267 | 0.04 | 0.004–0.540 | 0.015 |
| CBS (rs234706) | 1.90 | 0.798–4.533 | 0.147 | 95.12 | 3.455–2618 | 0.007 |
| APOE | 2.07 | 1.196–3.592 | 0.009 | 4.46 | 1.193–16.66 | 0.026 |
| _cons | 0.00 | 5.3 × 10−8–0.004 | 0.006 | |||
| SNPs | Dominant | Recessive | Additive | |||
|---|---|---|---|---|---|---|
| OR | p-Value | OR | p-Value | OR | p-Value | |
| Nervous system related | ||||||
| COMT (rs4633) | 22.05 | 0.017 | 3.41 | 0.123 | 5.50 | 0.014 |
| COMT (rs4680) | 1.00 | (omitted) | 1.00 | (omitted) | 1.00 | (omitted) |
| CYP2D6 (rs16947) | 14.92 | 0.044 | 3.22 | 0.076 | 3.64 | 0.030 |
| ADRB1 (rs1801253) | 15.51 | 0.013 | 1.00 | (omitted) | 9.61 | 0.009 |
| _cons | 0.003 | 0.009 | 0.39 | 0.050 | 0.00 | 0.003 |
| Methylation related | ||||||
| MTHFD1 (rs1076991) | 6.71 | 0.125 | 8.22 | 0.010 | 483.8 | 0.026 |
| MTHFD1 (rs2236225) | 2.19 | 0.416 | 3.28 | 0.149 | 22.22 | 0.036 |
| MAT1A (rs1985908) | 0.50 | 0.409 | 1.3 × 10−7 | 0.991 | 0.073 | 0.034 |
| CBS (rs234706) | 4.92 | 0.071 | 2.3 × 106 | 0.992 | 465.8 | 0.025 |
| APOE | 20.45 | <0.0001 | 1.00 | (omitted) | 161.3 | 0.022 |
| _cons | 0.018 | 0.033 | 0.29 | 0.017 | 1.9 × 10−7 | 0.024 |
| Mann Whitney, p < 0.05 (p ≤ 0.01 in bold) | Chi squared (R+Y), p ≤ 0.05 | Chi squared (Y+G), p ≤ 0.05 | EFFECT | |||||
|---|---|---|---|---|---|---|---|---|
| Protein | Genes (variant) | p value | Genes | p value | Genes | p value | AD | N |
| Apolipoprotein E4 fat metabolism—principle cholesterol carrier in brain supplying neurones via lipoprotein receptors | APOe4 | 1.61 × 10−6 | APOe4 | 0.029096332 | APOe4 | 4.12 × 10−32 | ||
| MethyleneTHF dehydrogenase long pathway replenishment of 5mTHF | MTHFD1 (rs1076991) | 0.000982 | MTHFD1 (rs1076991) | 0.010097315 | MTHFD1 (rs1076991) | 4.88 × 10−8 | ||
| MTHFD1 (rs2236225) | 0.000311491 | |||||||
| MethyleneTHF reductase final step in long and short pathway back to 5mTHF | MTHFR (rs1801131) | 0.026992 | ||||||
| synaptic vescile associated monoamine transporter | SLC18A1 (rs1390938) | 0.029941 | SLC18A1 (rs1390938) | 0.004797 | ||||
| monoamine transporter responsible for reuptake from synapse | SLC6A2 (rs5569) | 0.045301 | SLC6A2 (rs5569) | 0.01562887 | SLC6A2 (rs5569) | 0.015629 | ||
| Cytochrome oxidase involved in metabolism of xenobiotics | CYP2D6 (rs1135840) | 0.036104 | CYP2C19 (rs4244285) | 0.029096332 | ||||
| Mitochondrial enzyme—sulfite oxidase—detox | SUOX (rs705703) | 0.00987 | SUOX (rs705703) | 0.012419331 | ||||
| β-Adrenergic receptor | ADRB1 (rs1801253) | 0.032407 | ADRB1 (rs1801253) | 0.010097 | ||||
| Catechol-O-methyl transferase—degrades monoamines | COMT (rs4633) | 0.002408 | COMT (rs4633) | 2.15×10−5 | COMT (rs4633) | 0.001832 | ||
| COMT (rs4680) | 0.005641 | COMT (rs4680) | 0.001155233 | COMT (rs4680) | 0.008119 | |||
| cytochrome P450 Breakdown of medicines | CYP2D6 (rs16947) | 0.0107 | CYP2D6 (rs16947) | 9.00×10−5 | CYP2D6 (rs1135840) | 0.001063 | ||
| Iodothyronine deiodinase activates thyroid hormone | DIO2 (rs225014) | 0.045327562 | DIO2 (rs225014) | 0.026992 | ||||
| superoxide dismutase—detox from oxidative products | SOD2 (rs2758331) | 0.004987 | SOD2 (rs2758331) | 0.000221847 | SOD2 (rs2758331) | 0.004267 | ||
| SOD2 (rs4880) | 0.009887 | SOD2 (rs4880) | 0.000221847 | SOD2 (rs4880) | 0.014306 | |||
| Glutathione S-transferase—detox from drugs, environmental toxins, oxidative stress | GSTM1 (insert/delete) | 0.035014981 | GSTM1 (insert/delete) | 0.035015 | ||||
| Monoamine oxidase | MAOA (rs6323) | 0.019208 | ||||||
| 5HT receptor 2A | 5-HT2A (rs6311) | 0.002088939 | ||||||
| Dopamine receptor D2 | DRD2 (rs6277) | 0.045500264 | ||||||
| IFN-g (rs2430561) | 0.025935446 | |||||||
| iodothyronine deiodinase deiodination of T4 | DIO1 (rs2235544) | 0.012419331 | ||||||
| Solute carrier—high affinity transport of organic anions (e.g. T4 and other hormones) may act at BBB | SLCO1C1 (rs10770704) | 0.002199647 | ||||||
| Betaine--homocysteine S-methyltransferase 1 required for Hcyst to Methionine | BHMT (rs567754) | 0.043951044 | ||||||
| Cystathionine beta-synthase downregulates methionine by converting HCYst to cycsteine | CBS (rs234706) | 0.045327562 | ||||||
| Glutathione S-transferase P-conjugates glutathione to wide range of electrophiles/toxins | GSTP1 (rs1695) | 0.041226833 | ||||||
| Normal Ageing | Alzheimer’s | Normal Controls | Alzheimer’s Controls | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SampleIDs Target molecules | 12_11 | 09_31 | 14_09 | 14_11 | 12_01 | 13_45 | 14_50 | 19_29 | 19_31 | 11_25 | 14_08 | 17_36 | 17_34 | 19_04 A | 12_05 B | 12_32 C | 11_28 D |
| MTHFD1 (rs1076991) | CT | TT | TT | TT | TT | TT | TT | TT | TT | CC | CC | CT | CC | CT | CT | CT | CT |
| MTHFD1 (rs2236225) | AA | AG | AA | AA | AA | AA | GG | GG | AA | GG | GG | AG | AG | AG | GG | GG | |
| MTHFR (rs1801131) | TT | GT | GT | TT | GG | GG | TT | GG | TT | GT | GT | TT | TT | TT | GT | GT | GT |
| MTHFR (rs1801133) | AA | GG | AG | AG | GG | GG | AA | GG | GG | AG | AG | GG | GG | AG | AG | GG | GG |
| DOT BLOTS | |||||||||||||||||
| Homocysteine | 9190 | 10,600 | 5850 | 10,900 | 14,800 | 12,900 | 6640 | 7230 | 21,100 | 8490 | 26,100 | 3510 | 9720 | 14,700 | 49,000 | 18,500 | 157 |
| SAM | 7010 | 27,700 | 8080 | 23,100 | 9330 | 16,800 | 15,500 | 18,200 | 27,000 | 6000 | 38,000 | 8040 | 28,000 | 11,300 | 68,900 | 32,100 | 27,500 |
| Glutathione | 58,600 | 90,200 | 91,400 | 78,100 | 97,600 | 69,100 | 160,000 | 119,000 | 97,400 | 25,000 | 60,600 | 39,000 | 43,200 | 43,800 | 95,200 | 36,900 | 63,800 |
| Folates | 75,800 | 44,100 | 80,800 | 60,400 | 34,800 | 32,800 | 88,700 | 40,400 | 50,600 | 19,600 | 44,700 | 46,200 | 49,700 | 46,000 | 99,200 | 31,400 | 35,100 |
| WESTERN BLOTS | |||||||||||||||||
| MTHFD1 | 8560 | 9910 | 2070 | 9990 | 5390 | 5490 | 4530 | 3360 | 4680 | 873 | 3540 | 6200 | 7650 | 19,700 | 9210 | 36000 | 28,500 |
| MTHFR | NU | NU | 4850 | 15,000 | 2690 | 3130 | 5110 | 6980 | 6030 | 4850 | 4420 | 5290 | 5630 | 14,100 | 3470 | 7200 | 6650 |
| MTR | NU | NU | 2900 | 3950 | 2020 | 2840 | 2380 | 1570 | 2120 | 1230 | 863 | 717 | 452 | 5290 | 4260 | 2740 | 2290 |
| p Values | |||
|---|---|---|---|
| CvN | CvA | CvAC | |
| Hcyst | 0.554 | 0.915 | 0.303 |
| SAM | 0.267 | 0.737 | 0.139 |
| Glut | 0.005 | 0.008 | 0.098 |
| MTHFD1 | 0.332 | 0.931 | 0.033 |
| MTHFR | 0.530 | 0.795 | 0.297 |
| MTR | 0.145 | 0.002 | 0.013 |
| Folate | 0.140 | 0.497 | 0.502 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyan, J.; Buttercase, C.; Beswick, E.; Miyan, S.; Moshkdanian, G.; Naz, N. Folate Related Pathway Gene Analysis Reveals a Novel Metabolic Variant Associated with Alzheimer’s Disease with a Change in Metabolic Profile. Metabolites 2022, 12, 475. https://doi.org/10.3390/metabo12060475
Miyan J, Buttercase C, Beswick E, Miyan S, Moshkdanian G, Naz N. Folate Related Pathway Gene Analysis Reveals a Novel Metabolic Variant Associated with Alzheimer’s Disease with a Change in Metabolic Profile. Metabolites. 2022; 12(6):475. https://doi.org/10.3390/metabo12060475
Chicago/Turabian StyleMiyan, Jaleel, Charlotte Buttercase, Emma Beswick, Salma Miyan, Ghazaleh Moshkdanian, and Naila Naz. 2022. "Folate Related Pathway Gene Analysis Reveals a Novel Metabolic Variant Associated with Alzheimer’s Disease with a Change in Metabolic Profile" Metabolites 12, no. 6: 475. https://doi.org/10.3390/metabo12060475
APA StyleMiyan, J., Buttercase, C., Beswick, E., Miyan, S., Moshkdanian, G., & Naz, N. (2022). Folate Related Pathway Gene Analysis Reveals a Novel Metabolic Variant Associated with Alzheimer’s Disease with a Change in Metabolic Profile. Metabolites, 12(6), 475. https://doi.org/10.3390/metabo12060475