
NPvis: An Interactive Visualizer of Peptidic Natural Product–MS/MS Matches



Abstract
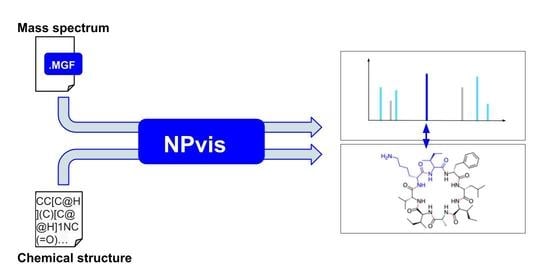
:
{kind=link}
{kind=link}
1. Introduction
2. Results
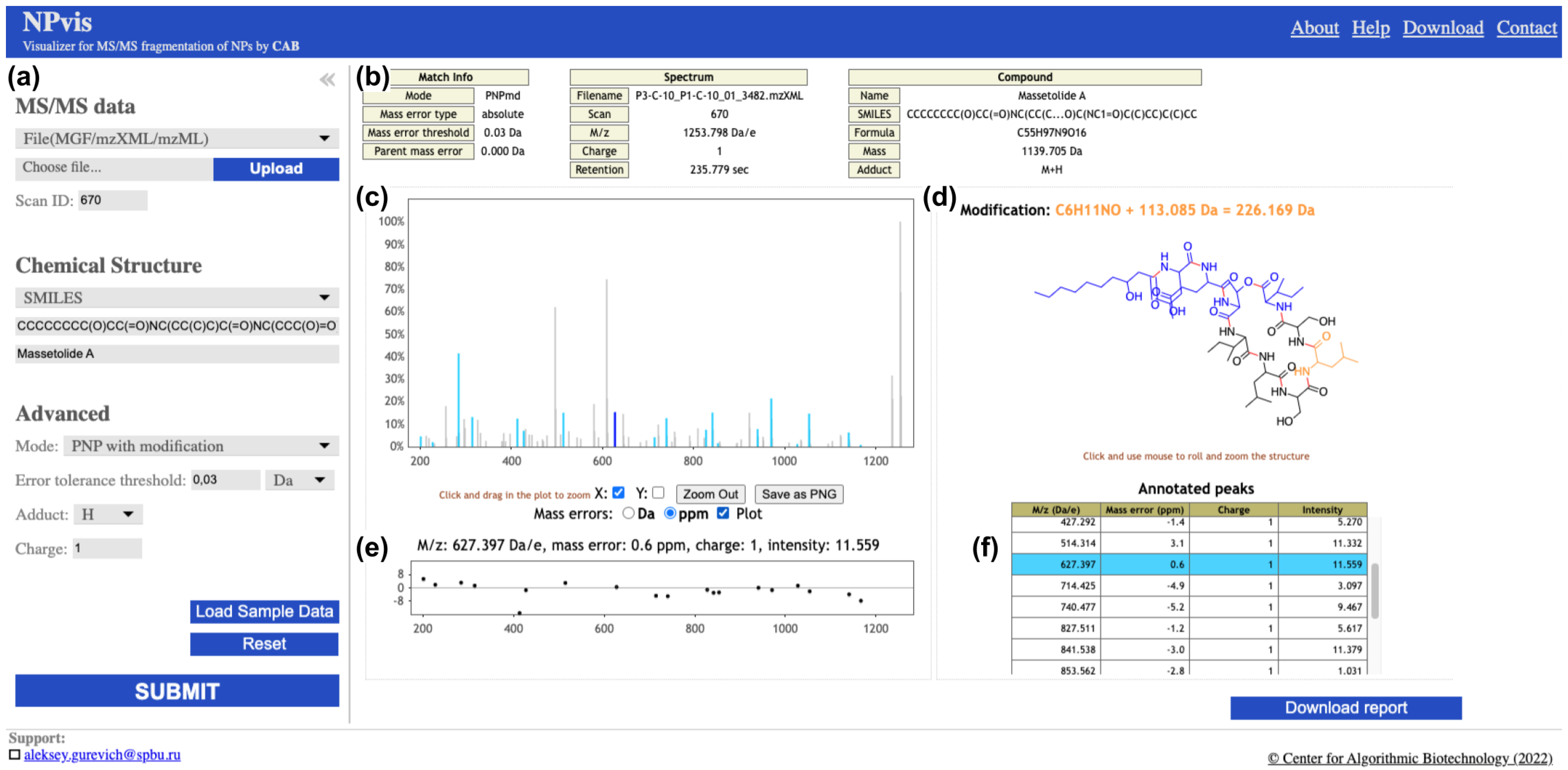
2.1. NPvis Overview
2.2. Web Server Design and Features
2.3. Integration with Metabolomics Platforms
3. Discussion
4. Materials and Methods
4.1. Pipeline and Web Server Implementation
4.2. MS/MS Fragmentation Model and Identification of Modifications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Acharya, D.; Adholeya, A.; Barrow, C.J.; Deshmukh, S.K. Nonribosomal peptides from marine microbes and their antimicrobial and anticancer potential. Front. Pharmacol. 2017, 8, 828. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.L.; Zhang, A.H.; Kong, L.; Wang, X.J. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv. 2018, 8, 22335–22350. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, L.; Shi, X.; Xu, G. New advances in analytical methods for mass spectrometry-based large-scale metabolomics study. TrAC Trends Anal. Chem. 2019, 121, 115665. [Google Scholar] [CrossRef]
- Heiles, S. Advanced tandem mass spectrometry in metabolomics and lipidomics—Methods and applications. Anal. Bioanal. Chem. 2021, 413, 5927–5948. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, W.; Yang, J.; Wang, H.; Qian, K. Nanoparticle-assisted cation adduction and fragmentation of small metabolites. Angew. Chem. Int. Ed. 2021, 60, 11310–11317. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2017, 13, 30–37. [Google Scholar] [CrossRef]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI: FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef]
- Ricart, E.; Pupin, M.; Muller, M.; Lisacek, F. Automatic annotation and dereplication of tandem mass spectra of peptidic natural products. Anal. Chem. 2020, 92, 15862–15871. [Google Scholar] [CrossRef]
- Li, K.; Vaudel, M.; Zhang, B.; Ren, Y.; Wen, B. PDV: An integrative proteomics data viewer. Bioinformatics 2019, 35, 1249–1251. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Carlson, A.; Sinitcyn, P.; Mann, M.; Cox, J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 2015, 15, 1453–1456. [Google Scholar] [CrossRef]
- Wang, R.; Fabregat, A.; Ríos, D.; Ovelleiro, D.; Foster, J.M.; Côté, R.G.; Griss, J.; Csordas, A.; Perez-Riverol, Y.; Reisinger, F.; et al. PRIDE Inspector: A tool to visualize and validate MS proteomics data. Nat. Biotechnol. 2012, 30, 135–137. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef]
- Stolze, S.C.; Kaiser, M. Challenges in the syntheses of peptidic natural products. Synthesis 2012, 44, 1755–1777. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Gurevich, A.; Mikheenko, A.; Shlemov, A.; Korobeynikov, A.; Mohimani, H.; Pevzner, P.A. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat. Microbiol. 2018, 3, 319–327. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; De Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Perez-Riverol, Y.; Carver, J.; Kawano, S.; Mendoza, L.; Van Den Bossche, T.; Gabriels, R.; Binz, P.A.; Pullman, B.; Sun, Z.; et al. Universal Spectrum Identifier for mass spectra. Nat. Methods 2021, 18, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Guler, M.; Tagirdzhanov, A.; Lee, Y.Y.; Gurevich, A.; Mohimani, H. MolDiscovery: Learning mass spectrometry fragmentation of small molecules. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Django: A Web Framework for the Python Programming Language. Available online: https://www.djangoproject.com/ (accessed on 15 June 2022).
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Chen, C.; Dorrestein, P.C.; Schymanski, E.L.; Schulze, T.; Neumann, S.; Meier, R.; Rogers, S.; Wang, M. Universal MS/MS visualization and retrieval with the metabolomics spectrum resolver web service. bioRxiv 2020. [Google Scholar] [CrossRef]
- Huber, F.; Verhoeven, S.; Meijer, C.; Spreeuw, H.; Castilla, E.M.V.; Geng, C.; van der Hooft, J.J.; Rogers, S.; Belloum, A.; Diblen, F.; et al. matchms-processing and similarity evaluation of mass spectrometry data. J. Open Source Softw. 2020, 5, 2411. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 15 June 2022).
- Burger, M.C. ChemDoodle Web Components: HTML5 toolkit for chemical graphics, interfaces, and informatics. J. Cheminformatics 2015, 7, 1–7. [Google Scholar] [CrossRef]
- Tagirdzhanov, A.M.; Shlemov, A.; Gurevich, A. NPS: Scoring and evaluating the statistical significance of peptidic natural product–spectrum matches. Bioinformatics 2019, 35, i315–i323. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunyavskaya, O.; Mikheenko, A.; Gurevich, A. NPvis: An Interactive Visualizer of Peptidic Natural Product–MS/MS Matches. Metabolites 2022, 12, 706. https://doi.org/10.3390/metabo12080706
Kunyavskaya O, Mikheenko A, Gurevich A. NPvis: An Interactive Visualizer of Peptidic Natural Product–MS/MS Matches. Metabolites. 2022; 12(8):706. https://doi.org/10.3390/metabo12080706
Chicago/Turabian StyleKunyavskaya, Olga, Alla Mikheenko, and Alexey Gurevich. 2022. "NPvis: An Interactive Visualizer of Peptidic Natural Product–MS/MS Matches" Metabolites 12, no. 8: 706. https://doi.org/10.3390/metabo12080706
APA StyleKunyavskaya, O., Mikheenko, A., & Gurevich, A. (2022). NPvis: An Interactive Visualizer of Peptidic Natural Product–MS/MS Matches. Metabolites, 12(8), 706. https://doi.org/10.3390/metabo12080706