
Convolutional Neural Network-Based Compound Fingerprint Prediction for Metabolite Annotation

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Evaluation of the CNN’s Performance in Metabolite Annotation
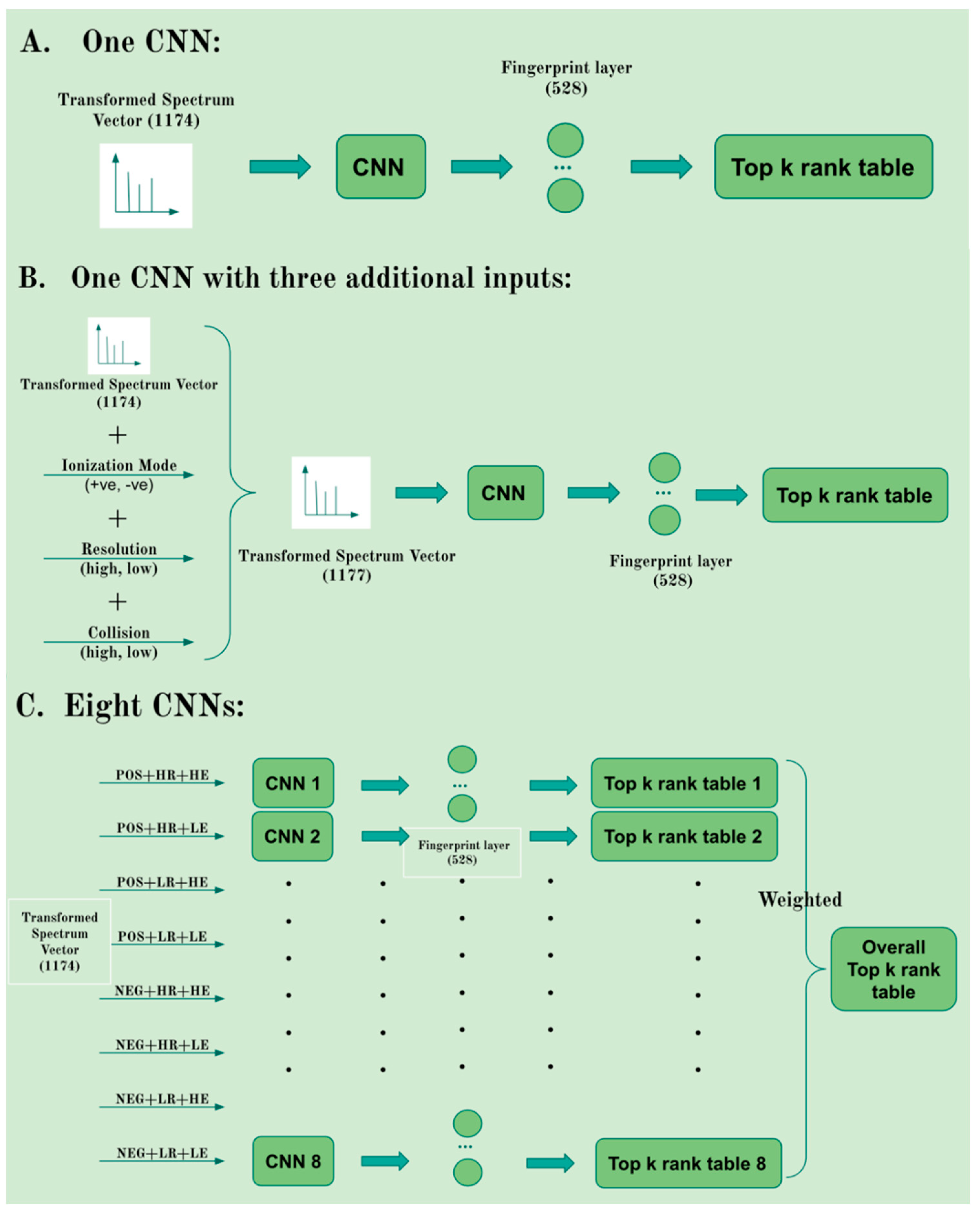
2.2. Evaluation of Single vs. Multiple CNNs
2.3. Evaluation of MetFID’s Performance Compared to Other Tools
3. Discussion
4. Materials and Methods
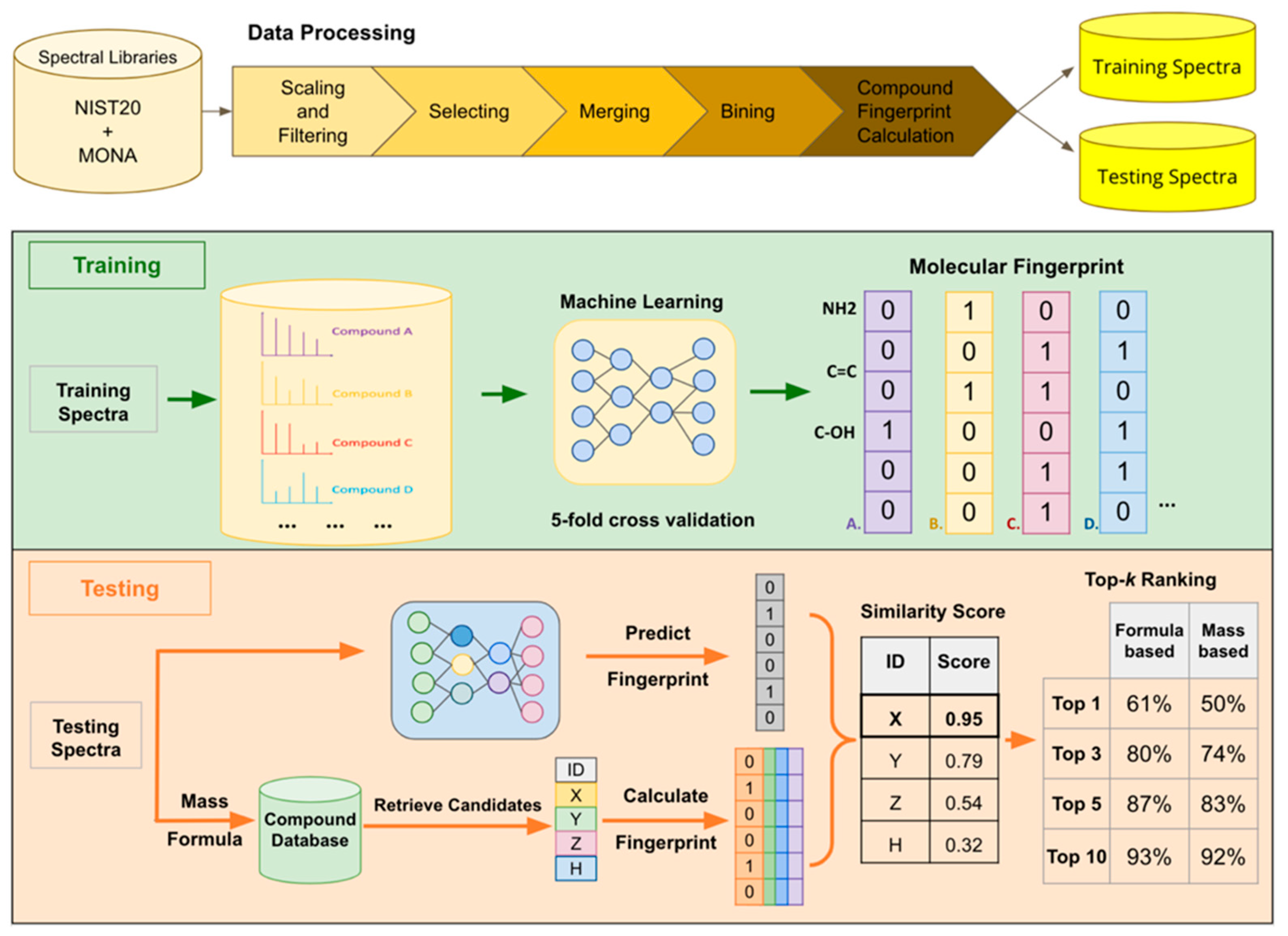
4.1. Workflow Overview
4.2. Data Processing
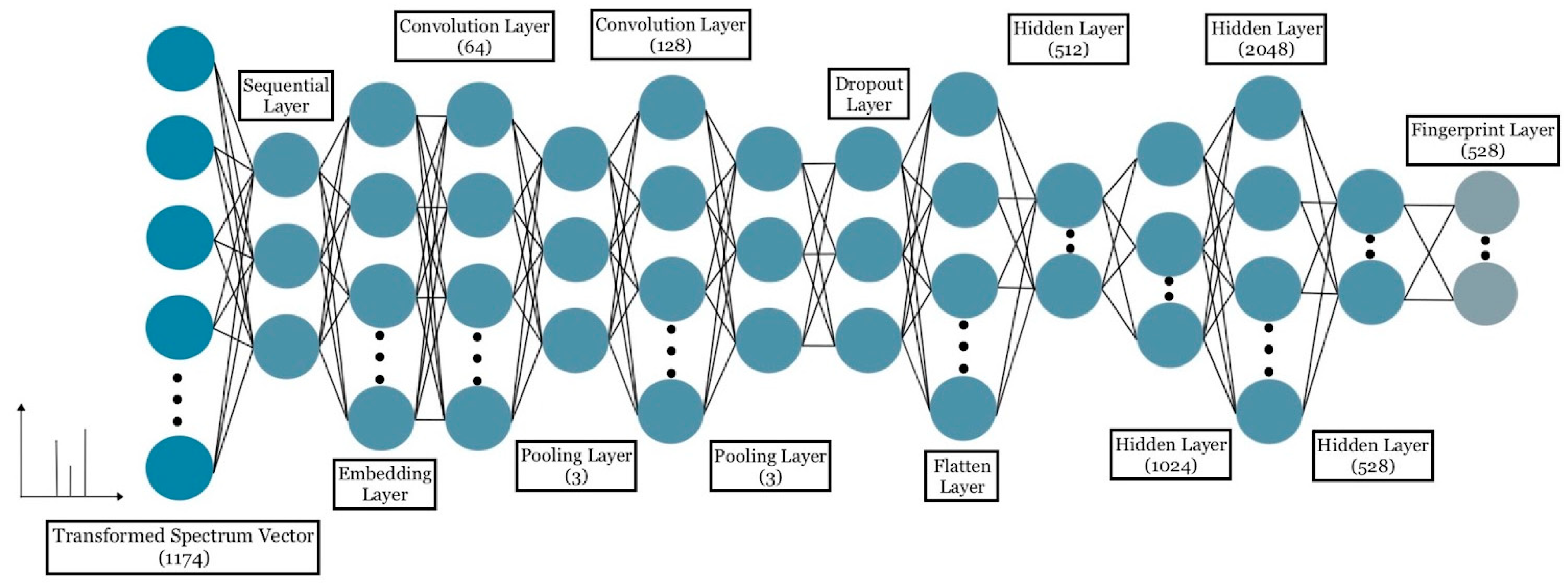
4.3. Training
4.4. Testing
4.5. Single vs. Multiple CNN Models
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Mathema, V.B.; Duangkumpha, K.; Wanichthanarak, K.; Jariyasopit, N.; Dhakal, E.; Sathirapongsasuti, N.; Kitiyakara, C.; Sirivatanauksorn, Y.; Khoomrung, S. CRISP: A deep learning architecture for GC × GC–TOFMS contour ROI identification, simulation and analysis in imaging metabolomics. Briefings Bioinform. 2022, 23, bbab550. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Lamichhane, S.; Mathema, V.B.; McGlinchey, A.; Dickens, A.M.; Khoomrung, S.; Orešič, M. Deep learning meets metabolomics: A methodological perspective. Briefings Bioinform. 2020, 22, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Nguyen, C.H.; Mamitsuka, H. Recent advances and prospects of computational methods for metabolite identification: A review with emphasis on machine learning approaches. Briefings Bioinform. 2018, 20, 2028–2043. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Central J. 2008, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaría, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of deep learning: Concepts, CNN architectures, challenges, applications, future directions. J. Big Data 2021, 8, 1–74. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Nguyen, C.H.; Mamitsuka, H. SIMPLE: Sparse Interaction Model over Peaks of moLEcules for fast, interpretable metabolite identification from tandem mass spectra. Bioinformatics 2018, 34, i323–i332. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2012, 9, 44–66. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Van Treuren, W.; Fischer, C.R.; Merrill, B.D.; DeFelice, B.C.; Sanchez, J.M.; Higginbottom, S.K.; Guthrie, L.; Fall, L.A.; Dodd, D.; et al. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 2021, 595, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Alley, A.; Ghaffari, K.; Ressom, H.W. MetFID: Artificial neural network-based compound fingerprint prediction for metabolite annotation. Metabolomics 2020, 16, 104. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.; Verma, G.K. Convolutional neural network: A review of models, methodologies and applications to object detection. Prog. Artif. Intell. 2019, 9, 85–112. [Google Scholar] [CrossRef]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, T.; Xu, J.; Luo, X.; Ying, Y. DeepSpectra: An end-to-end deep learning approach for quantitative spectral analysis. Anal. Chim. Acta 2019, 1058, 48–57. [Google Scholar] [CrossRef]
- Kim, H.W.; Zhang, C.; Cottrell, G.W.; Gerwick, W.H. SMART-Miner: A convolutional neural network-based metabolite identification from 1H-13C HSQC spectra. Org. Magn. Reson. 2021. [Google Scholar] [CrossRef]
- Fedorova, E.S.; Matyushin, D.D.; Plyushchenko, I.V.; Stavrianidi, A.N.; Buryak, A.K. Deep learning for retention time prediction in reversed-phase liquid chromatography. J. Chromatogr. A 2021, 1664, 462792. [Google Scholar] [CrossRef]
- Ji, H.; Deng, H.; Lu, H.; Zhang, Z. Predicting a Molecular Fingerprint from an Electron Ionization Mass Spectrum with Deep Neural Networks. Anal. Chem. 2020, 92, 8649–8653. [Google Scholar] [CrossRef]
- Li, D.-W.; Hansen, A.L.; Yuan, C.; Bruschweiler-Li, L.; Brüschweiler, R. DEEP picker is a deep neural network for accurate deconvolution of complex two-dimensional NMR spectra. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Laponogov, I.; Sadawi, N.; Galea, D.; Mirnezami, R.; Veselkov, K.A. ChemDistiller: An engine for metabolite annotation in mass spectrometry. Bioinformatics 2018, 34, 2096–2102. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Ghaffari, K.; Alley, A.; Ressom, H.W. Metabolite Identification Using Artificial Neural Network. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; IEEE: San Diego, CA, USA, 2019; pp. 244–248. [Google Scholar] [CrossRef]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf. Fusion 2018, 50, 71–91. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| LR | SLP | SVM | MLP | CNN | ||||||
| F1 | 61% | 59% | 66% | 67% | 71% | |||||
| Tanimoto | 45% | 43% | 52% | 53% | 58% | |||||
| Mass-Based | Formula-Based | |||||||||
| Rank | LR1 | SLP | SVM | MLP | CNN | LR | SLP | SVM | MLP | CNN |
| Top 1 | 32% | 35% | 39% | 40% | 43% | 45% | 47% | 49% | 48% | 50% |
| Top 3 | 59% | 61% | 66% | 66% | 69% | 68% | 68% | 70% | 71% | 71% |
| Top 5 | 71% | 72% | 75% | 75% | 77% | 75% | 75% | 77% | 77% | 78% |
| Top 10 | 81% | 82% | 83% | 82% | 84% | 81% | 82% | 82% | 81% | 82% |
| Dataset | CASMI 2016 | |||||||
| Libraries | NIST 20 + MoNA | |||||||
| SLP | SVM | MLP | CNN | |||||
| F1 | 49% | 52% | 53% | 56% | ||||
| Tanimoto | 33% | 36% | 38% | 41% | ||||
| Mass-Based | Formula-Based | |||||||
| Rank | SLP | SVM | MLP | CNN | SLP | SVM | MLP | CNN |
| Top 1 | 35% | 32% | 48% | 52% | 60% | 59% | 68% | 71% |
| Top 3 | 61% | 59% | 71% | 76% | 81% | 81% | 87% | 88% |
| Top 5 | 78% | 75% | 83% | 87% | 88% | 86% | 90% | 91% |
| Top 10 | 93% | 89% | 93% | 95% | 94% | 92% | 94% | 95% |
| CASMI 2016 Testing | ||||
|---|---|---|---|---|
| Mass-Based | Formula-Based | |||
| Rank | ChemDistiller | MetFID | CSI:FingerID | MetFID |
| Top 1 | 34% (44%) | 52% (53%) | 67% (73%) | 71% (74%) |
| Top 3 | 47% (59%) | 76% (76%) | 71% (78%) | 88% (91%) |
| Top 5 | 58% (73%) | 87% (87%) | 72% (79%) | 91% (94%) |
| Top 10 | 63% (80%) | 95% (96%) | 72% (79%) | 95% (99%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Chau, H.Y.K.; Wang, K.; Ao, H.; Varghese, R.S.; Ressom, H.W. Convolutional Neural Network-Based Compound Fingerprint Prediction for Metabolite Annotation. Metabolites 2022, 12, 605. https://doi.org/10.3390/metabo12070605
Gao S, Chau HYK, Wang K, Ao H, Varghese RS, Ressom HW. Convolutional Neural Network-Based Compound Fingerprint Prediction for Metabolite Annotation. Metabolites. 2022; 12(7):605. https://doi.org/10.3390/metabo12070605
Chicago/Turabian StyleGao, Shijinqiu, Hoi Yan Katharine Chau, Kuijun Wang, Hongyu Ao, Rency S. Varghese, and Habtom W. Ressom. 2022. "Convolutional Neural Network-Based Compound Fingerprint Prediction for Metabolite Annotation" Metabolites 12, no. 7: 605. https://doi.org/10.3390/metabo12070605
APA StyleGao, S., Chau, H. Y. K., Wang, K., Ao, H., Varghese, R. S., & Ressom, H. W. (2022). Convolutional Neural Network-Based Compound Fingerprint Prediction for Metabolite Annotation. Metabolites, 12(7), 605. https://doi.org/10.3390/metabo12070605