
Whole Exome Sequencing Enhanced Imputation Identifies 85 Metabolite Associations in the Alpine CHRIS Cohort

 ,
,  ,
,  ,
,  , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
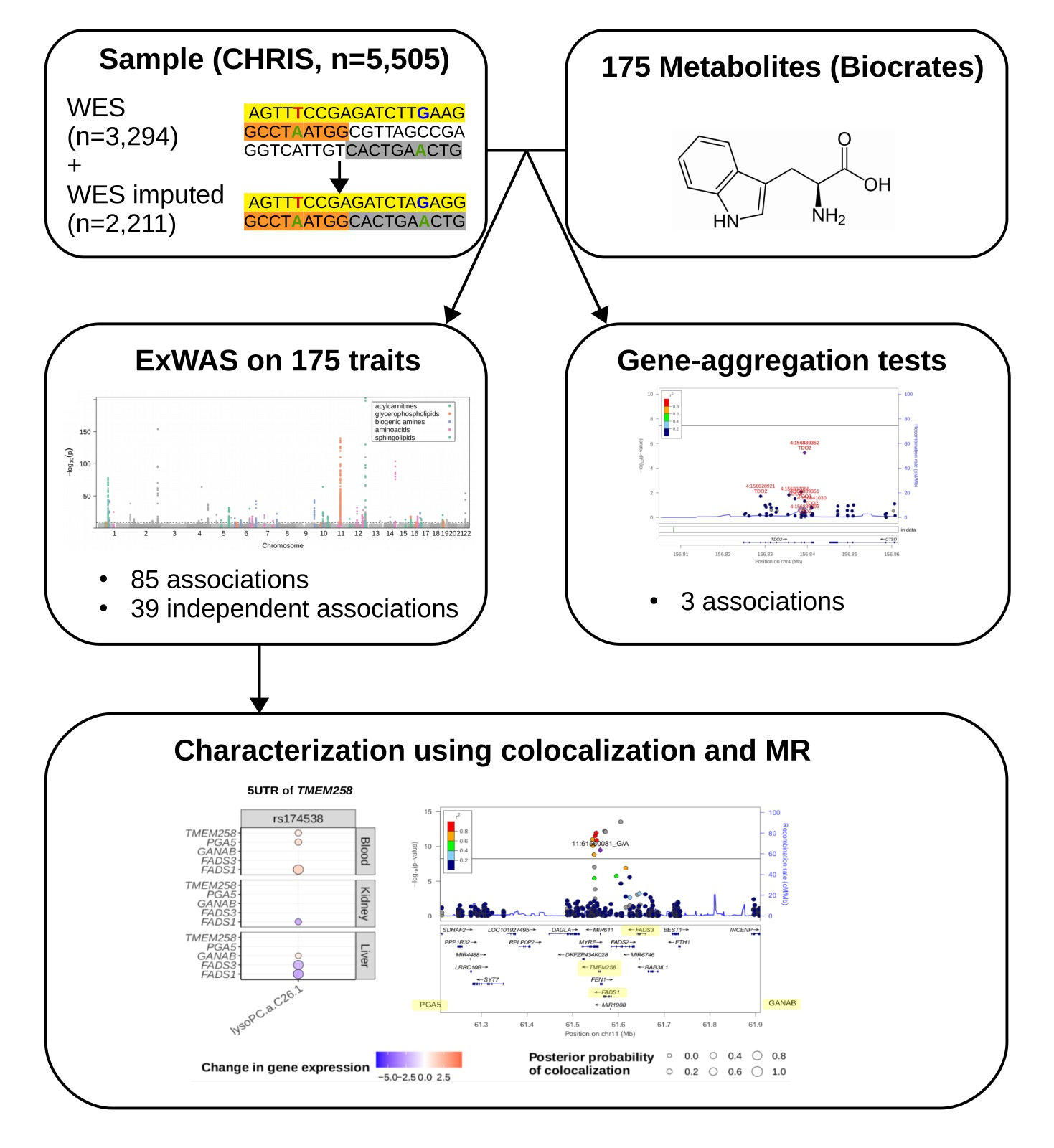
2.1. Study Design and Genotype Data
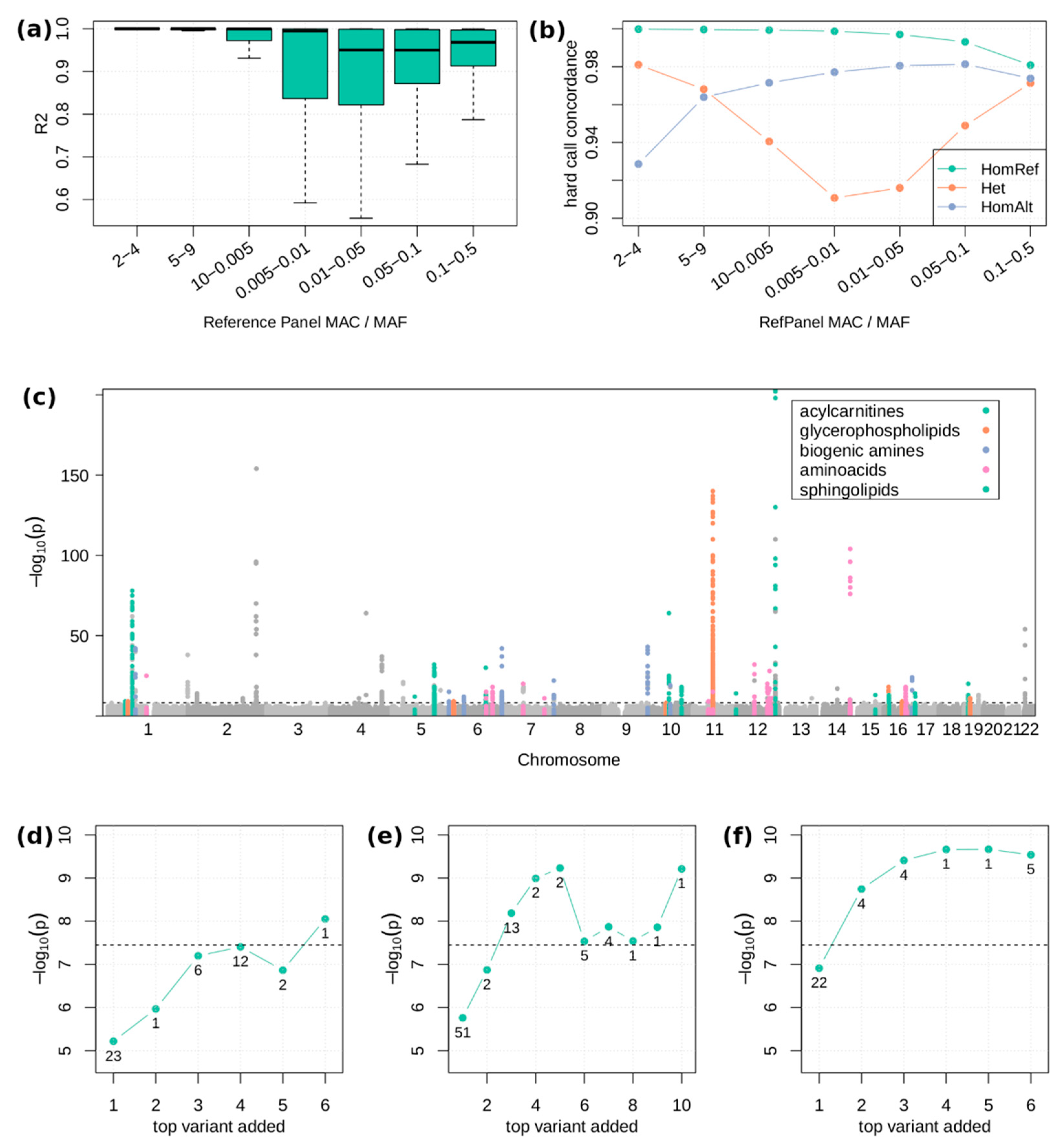
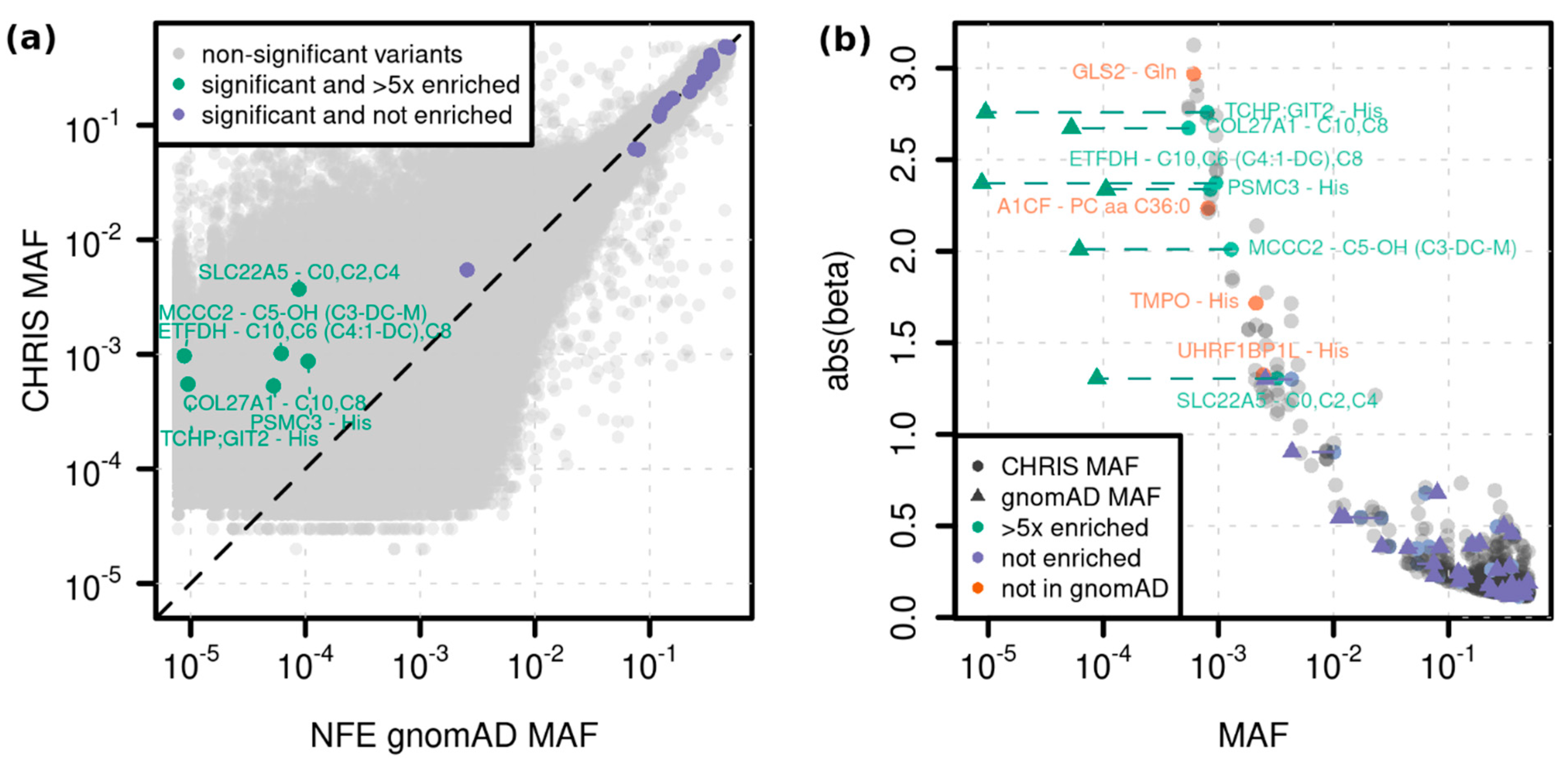
2.2. Single Variant Associations
2.3. Gene-Level Associations
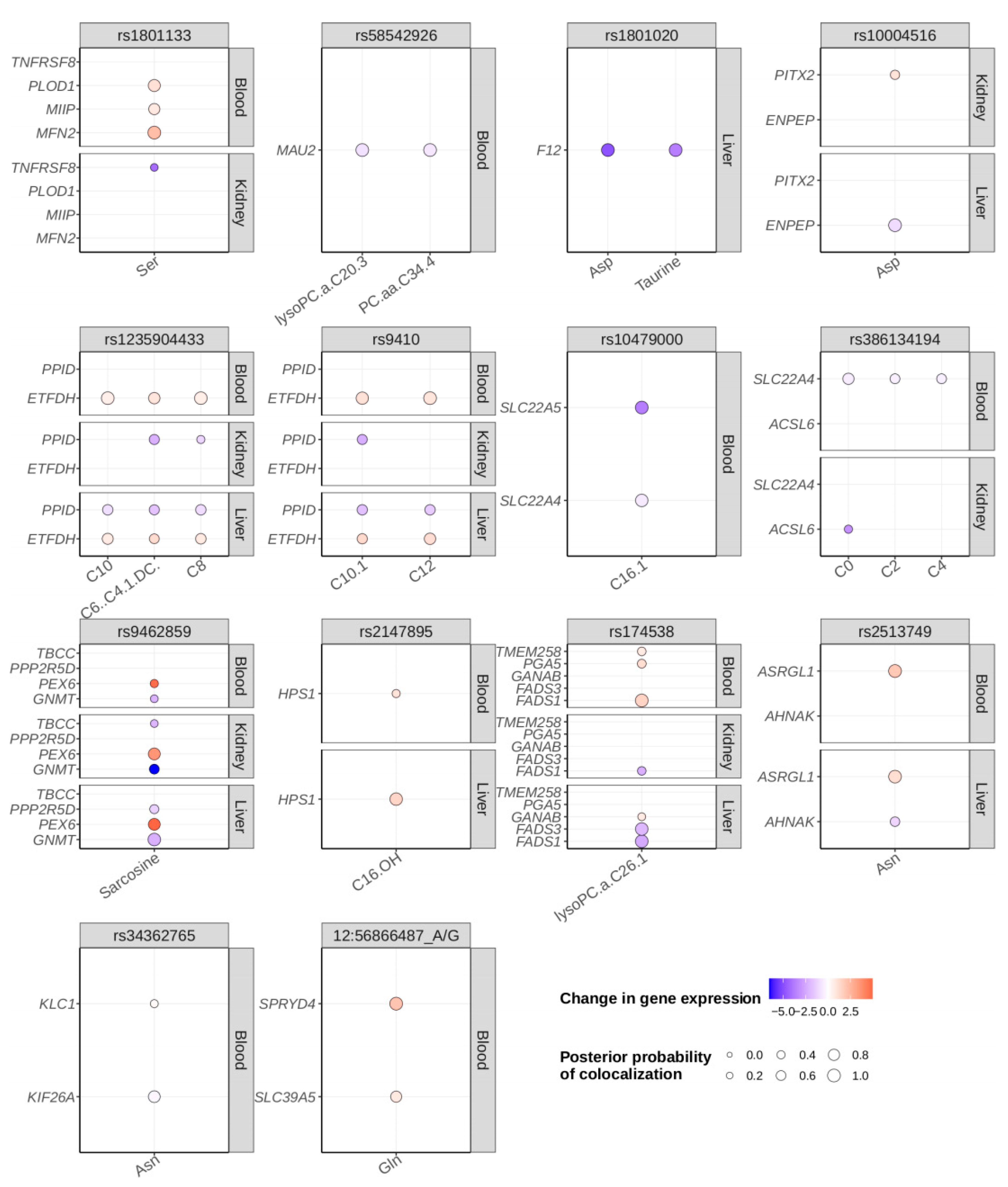
2.4. Colocalization Analysis
2.5. Mendelian Randomization
3. Discussion
4. Materials and Methods
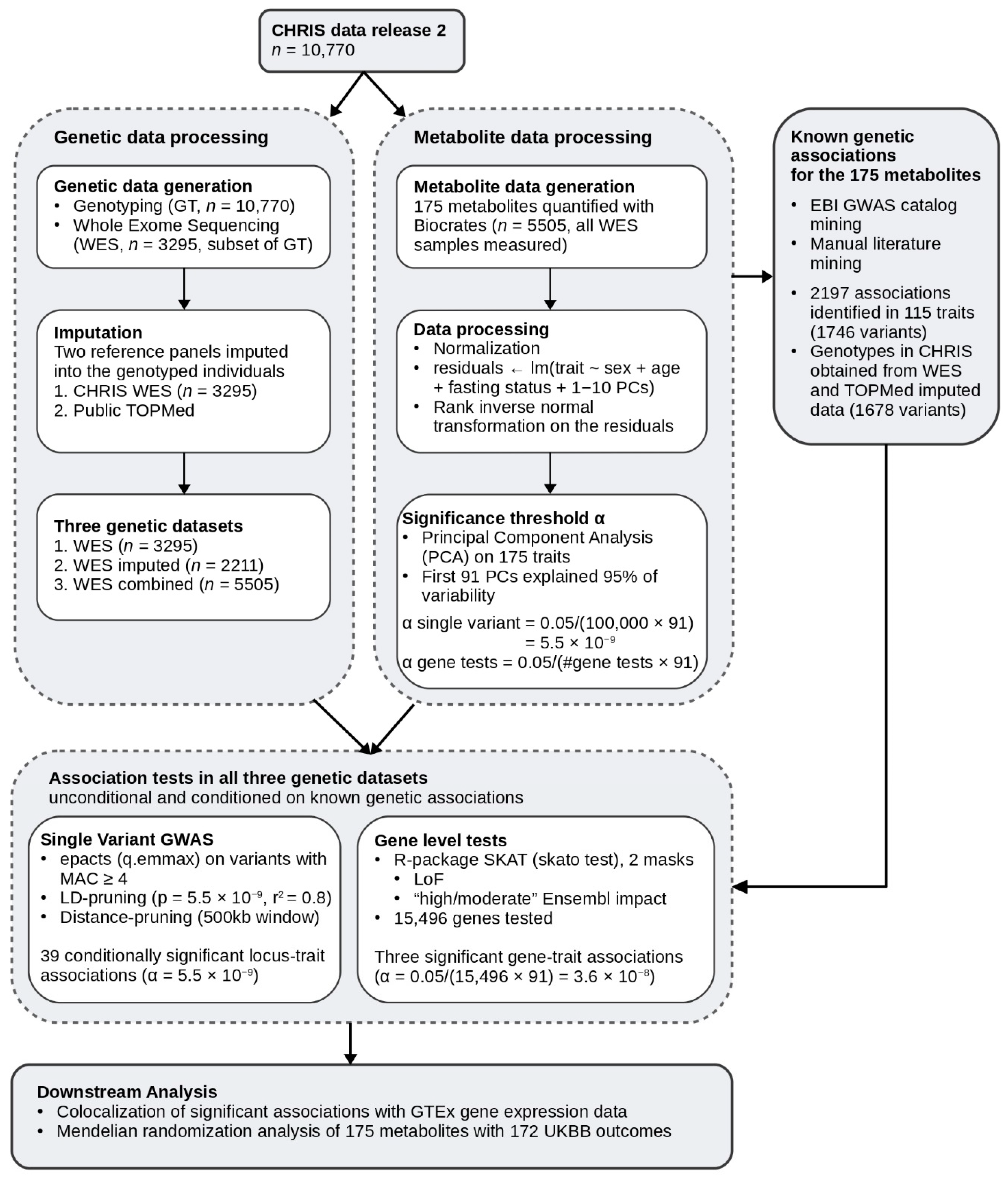
4.1. CHRIS Population Study
4.2. Genotyping
4.3. Whole Exome Sequencing
4.4. Genotype Imputation
4.5. Metabolomics Data
4.6. Definition of Datasets
- Whole exome sequencing (WES): All individuals with whole-exome sequencing and measured metabolite data (3294 individuals and 554,589 variants).
- Imputed only (WES imputed): All individuals with genotype data (and thereby imputed) that were not in the imputation reference panel with measured metabolite data, restricting to imputed variants only (2211 individuals 374,349 variants).
- Whole-exome sequencing combined with imputed (WES combined): All individuals with whole-exome sequencing data, genotype, and imputation data, and with measured metabolite data, combining sequenced, genotyped, and imputed variants (5505 individuals and 624,751 variants).
4.7. Known Genetic Associations and Conditional Analysis
4.8. Single Variant Association Tests
4.9. Gene Level Association Tests
4.10. Colocalization Analysis
4.11. Mendelian Randomization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Kastenmüller, G.; Raffler, J.; Gieger, C.; Suhre, K. Genetics of human metabolism: An update. Hum. Mol. Genet. 2015, 24, R93–R101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards Mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Aderemi, A.V.; Ayeleso, A.O.; Oyedapo, O.O.; Mukwevho, E. Metabolomics: A scoping review of its role as a tool for disease biomarker discovery in selected non-communicable diseases. Metabolites 2021, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Hagenbeek, F.A.; Pool, R.; van Dongen, J.; Draisma, H.H.M.; Jan Hottenga, J.; Willemsen, G.; Abdellaoui, A.; Fedko, I.O.; den Braber, A.; Visser, P.J.; et al. Heritability estimates for 361 blood metabolites across 40 genome-wide association studies. Nat. Commun. 2020, 11, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hysi, P.G.; Mangino, M.; Christofidou, P.; Falchi, M.; Karoly, E.D.; NIHR Bioresource Investigators; Mohney, R.P.; Valdes, A.M.; Spector, T.D.; Menni, C. Metabolome genome-wide association study identifies 74 novel genomic regions influencing plasma metabolites levels. Metabolites 2022, 12, 61. [Google Scholar] [CrossRef]
- Lotta, L.A.; Pietzner, M.; Stewart, I.D.; Wittemans, L.B.L.; Li, C.; Bonelli, R.; Raffler, J.; Biggs, E.K.; Oliver-Williams, C.; Auyeung, V.P.W.; et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet. 2021, 53, 54–64. [Google Scholar] [CrossRef]
- Nag, A.; Kurushima, Y.; Bowyer, R.C.E.; Wells, P.M.; Weiss, S.; Pietzner, M.; Kocher, T.; Raffler, J.; Völker, U.; Mangino, M.; et al. Genome-wide scan identifies novel genetic loci regulating salivary metabolite levels. Hum. Mol. Genet. 2020, 29, 864–875. [Google Scholar] [CrossRef]
- Schlosser, P.; Li, Y.; Sekula, P.; Raffler, J.; Grundner-Culemann, F.; Pietzner, M.; Cheng, Y.; Wuttke, M.; Steinbrenner, I.; Schultheiss, U.T.; et al. Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans. Nat. Genet. 2020, 52, 167–176. [Google Scholar] [CrossRef]
- Barton, A.R.; Sherman, M.A.; Mukamel, R.E.; Loh, P.-R. Whole-exome imputation within UK biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet. 2021, 53, 1260–1269. [Google Scholar] [CrossRef]
- Pattaro, C.; Gögele, M.; Mascalzoni, D.; Melotti, R.; Schwienbacher, C.; De Grandi, A.; Foco, L.; D’Elia, Y.; Linder, B.; Fuchsberger, C.; et al. The cooperative health research in south tyrol (CHRIS) study: Rationale, objectives, and preliminary results. J. Transl. Med. 2015, 13, 348. [Google Scholar] [CrossRef]
- Nag, A.; Middleton, L.; Dhindsa, R.S.; Vitsios, D.; Wigmore, E.; Allman, E.L.; Reznichenko, A.; Carss, K.; Smith, K.R.; Wang, Q.; et al. Assessing the contribution of rare-to-common protein-coding variants to circulating metabolic biomarker levels via 412,394 UK Biobank Exome Sequences. MedRxiv 2021. [Google Scholar] [CrossRef]
- Sahai, I.; Garganta, C.L.; Bailey, J.; James, P.; Levy, H.L.; Martin, M.; Neilan, E.; Phornphutkul, C.; Sweetser, D.A.; Zytkovicz, T.H.; et al. Newborn screening for glutaric aciduria-II: The new england experience. JIMD Rep. 2014, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, M.F.; Suormala, T.; Randolph, A.; Coelho, D.; Fowler, B.; Valle, D.; Baumgartner, M.R. 3-methylcrotonyl-CoA carboxylase deficiency: Mutation analysis in 28 probands, 9 symptomatic and 19 detected by newborn screening. Hum. Mutat. 2005, 26, 164. [Google Scholar] [CrossRef]
- Grünert, S.C.; Stucki, M.; Morscher, R.J.; Suormala, T.; Bürer, C.; Burda, P.; Christensen, E.; Ficicioglu, C.; Herwig, J.; Kölker, S.; et al. 3-methylcrotonyl-CoA carboxylase deficiency: Clinical, biochemical, enzymatic and molecular Studies in 88 individuals. Orphanet. J. Rare Dis. 2012, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Stadler, S.C.; Polanetz, R.; Maier, E.M.; Heidenreich, S.C.; Niederer, B.; Mayerhofer, P.U.; Lagler, F.; Koch, H.-G.; Santer, R.; Fletcher, J.M.; et al. Newborn screening for 3-methylcrotonyl-CoA carboxylase deficiency: Population heterogeneity of MCCA and MCCB mutations and impact on risk assessment. Hum. Mutat. 2006, 27, 748–759. [Google Scholar] [CrossRef]
- Kalcheva, I.D.; Matsuda, Y.; Plass, C.; Chapman, V.M. Isolation and characterization of a pseudoautosomal region-specific Genetic marker in C57BL/6 mice using genomic representational difference analysis. Proc. Natl. Acad. Sci. USA 1995, 92, 12352–12356. [Google Scholar] [CrossRef] [Green Version]
- Šimkovič, M.; Degala, G.D.; Eaton, S.S.; Frerman, F.E. Expression of human electron transfer flavoprotein-ubiquinone oxidoreductase from a baculovirus vector: Kinetic and spectral characterization of the human protein. Biochem. J. 2002, 364, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Draisma, H.H.M.; Pool, R.; Kobl, M.; Jansen, R.; Petersen, A.-K.; Vaarhorst, A.A.M.; Yet, I.; Haller, T.; Demirkan, A.; Esko, T.; et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat. Commun. 2015, 6, 7208. [Google Scholar] [CrossRef] [Green Version]
- Krumsiek, J.; Suhre, K.; Evans, A.M.; Mitchell, M.W.; Mohney, R.P.; Milburn, M.V.; Wägele, B.; Römisch-Margl, W.; Illig, T.; Adamski, J.; et al. Mining the unknown: A systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012, 8, e1003005. [Google Scholar] [CrossRef] [Green Version]
- Anant, S.; Blanc, V.; Davidson, N.O. Molecular regulation, evolutionary, and functional adaptations associated with C to U editing of mammalian apolipoproteinB MRNA. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2003; Volume 75, pp. 1–41. [Google Scholar]
- Ferreira, P.; Shin, I.; Sosova, I.; Dornevil, K.; Jain, S.; Dewey, D.; Liu, F.; Liu, A. Hypertryptophanemia Due to tryptophan 2,3-dioxygenase deficiency. Mol. Genet. Metab. 2017, 120, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A.A.-B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindseth, G.; Helland, B.; Caspers, J. The effects of dietary tryptophan on affective disorders. Arch. Psychiatr. Nurs. 2015, 29, 102–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, A.M.; Tanabe, A.; Iwahori, Y. A systematic review of the effect of L-tryptophan supplementation on mood and emotional functioning. J. Diet. Suppl. 2021, 18, 316–333. [Google Scholar] [CrossRef]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielberger, C.D. Manual for the State-Trait Anxietry, Inventory; Consulting Psychologist: Palo Alto, CA, USA, 1970. [Google Scholar]
- Codoñer-Franch, P.; Tavárez-Alonso, S.; Murria-Estal, R.; Herrera-Martín, G.; Alonso-Iglesias, E. Polyamines are increased in obese children and are related to markers of oxidative/nitrosative stress and angiogenesis. J. Clin. Endocrinol. Metab. 2011, 96, 2821–2825. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, J.C.; Delpino-Rius, A.; Samarra, I.; Castellano-Castillo, D.; Muñoz-Garach, A.; Bernal-Lopez, M.R.; Queipo-Ortuño, M.I.; Cardona, F.; Ramos-Molina, B.; Tinahones, F.J. Type 2 diabetes is associated with a different pattern of serum polyamines: A case−control study from the predimed-plus trial. J. Clin. Med. 2019, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Chan, L.S.; Bose, D.; Jackson, A.U.; VandeHaar, P.; Locke, A.E.; Fuchsberger, C.; Stringham, H.M.; Welch, R.; Yu, K.; et al. Genome-wide association studies of metabolites in finnish men identify disease-relevant loci. Nat. Commun. 2022, 13, 1644. [Google Scholar] [CrossRef]
- Cheng, Y.; Schlosser, P.; Hertel, J.; Sekula, P.; Oefner, P.J.; Spiekerkoetter, U.; Mielke, J.; Freitag, D.F.; Schmidts, M.; Kronenberg, F.; et al. Rare genetic variants affecting urine metabolite levels link population variation to inborn errors of metabolism. Nat. Commun. 2021, 12, 964. [Google Scholar] [CrossRef]
- Skol, A.D.; Scott, L.J.; Abecasis, G.R.; Boehnke, M. Joint analysis is more efficient than replication-based Analysis for two-stage genome-wide association studies. Nat. Genet. 2006, 38, 209–213. [Google Scholar] [CrossRef]
- Du, M.; Auer, P.L.; Jiao, S.; Haessler, J.; Altshuler, D.; Boerwinkle, E.; Carlson, C.S.; Carty, C.L.; Chen, Y.-D.I.; Curtis, K.; et al. Whole-exome imputation of sequence variants identified two novel alleles associated with adult body height in African Americans. Hum. Mol. Genet. 2014, 23, 6607–6615. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Liu, E.Y.; Auer, P.L.; Zhang, G.; Lange, E.M.; Jun, G.; Bizon, C.; Jiao, S.; Buyske, S.; Franceschini, N.; et al. Imputation of coding variants in African Americans: Better performance using data from the exome sequencing project. Bioinformatics 2013, 29, 2744–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, G.G.; Hall, I.M. Samblaster: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Li, B.; Zhan, X.; Wing, M.-K.; Anderson, P.; Kang, H.M.; Abecasis, G.R. QPLOT: A quality assessment tool for next generation sequencing data. Biomed. Res. Int. 2013, 2013, 865181. [Google Scholar] [CrossRef] [Green Version]
- Jun, G.; Flickinger, M.; Hetrick, K.N.; Romm, J.M.; Doheny, K.F.; Abecasis, G.R.; Boehnke, M.; Kang, H.M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 2012, 91, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified representation of genetic variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LOFTEE (Loss-of-Function Transcript Effect Estimator). Available online: https://github.com/konradjk/loftee (accessed on 29 April 2022).
- O’Connell, J.; Gurdasani, D.; Delaneau, O.; Pirastu, N.; Ulivi, S.; Cocca, M.; Traglia, M.; Huang, J.; Huffman, J.E.; Rudan, I.; et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 2014, 10, e1004234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Abecasis, G.R.; Browning, B.L. Genotype imputation from large reference panels. Annu. Rev. Genomics Hum. Genet. 2018, 19, 73–96. [Google Scholar] [CrossRef] [PubMed]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Verri Hernandes, V.; Dordevic, N.; Hantikainen, E.M.; Sigurdsson, B.B.; Smárason, S.V.; Garcia-Larsen, V.; Gögele, M.; Caprioli, G.; Bozzolan, I.; Pramstaller, P.P.; et al. Age, sex, body mass index, diet and menopause related metabolites in a large homogeneous alpine cohort. Metabolites 2022, 12, 205. [Google Scholar] [CrossRef]
- Fadista, J.; Manning, A.K.; Florez, J.C.; Groop, L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 2016, 24, 1202–1205. [Google Scholar] [CrossRef] [Green Version]
- Statgen/Swiss. Available online: https://github.com/statgen/swiss (accessed on 29 April 2022).
- Calculated Consequences. Available online: https://grch37.ensembl.org/info/genome/variation/prediction/predicted_data.html (accessed on 29 April 2022).
- SKAT: SNP-Set (Sequence) Kernel Association Test. Available online: https://cran.r-project.org/web/packages/SKAT/index.html (accessed on 29 April 2022).
- Tobyjohnson/Gtx. Available online: https://github.com/tobyjohnson/gtx (accessed on 29 April 2022).
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.P.; Davies, N.M.; Hemani, G.; Davey Smith, G. Two-sample mendelian randomization: Avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int. J. Epidemiol. 2016, 45, 1717–1726. [Google Scholar] [CrossRef] [Green Version]
- Study Design|Pan UKBB. Available online: https://pan-dev.ukbb.broadinstitute.org/docs/study-design (accessed on 29 April 2022).
- Li, B.; Martin, E.B. An approximation to the F distribution using the chi-square distribution. Comput. Stat. Data Anal. 2002, 40, 21–26. [Google Scholar] [CrossRef]
- Greco, M.F.D.; Minelli, C.; Sheehan, N.A.; Thompson, J.R. Detecting pleiotropy in mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 2015, 34, 2926–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid Instruments: Effect estimation and bias detection through egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments Using a weighted median estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data mendelian randomization via the zero Modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slob, E.A.W.; Burgess, S. A comparison of robust mendelian randomization methods using summary data. Genet. Epidemiol. 2020, 44, 313–329. [Google Scholar] [CrossRef] [Green Version]
- Pan-UK Biobank Phenotype Manifest—Google Sheets. Available online: https://docs.google.com/spreadsheets/d/1AeeADtT0U1AukliiNyiVzVRdLYPkTbruQSk38DeutU8/edit#gid=511623409 (accessed on 29 April 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait Code | Trait Name | Gene (LD Mapped Gene) | Variant (Rsid) | Effect | p-Value (Conditioned) | Beta (SE) | MAF |
|---|---|---|---|---|---|---|---|
| Ser | Serine | MTHFR | 1:11856378_G/A (rs1801133) | missense | 3.4 × 10−8 (4.0 × 10−9) | −0.11 (0.02) | 0.405 |
| Asp | Aspartate | ENPEP | 4:111398208_A/G (rs10004516) | missense | 1.2 × 10−7 (1.8 × 10−11) | 0.16 (0.03) | 0.127 |
| C10 | Decanoylcarnitine | ETFDH | 4:159618737_G/A (rs1235904433) | stop gained | 1.1 × 10−12 (2.5 × 10−16) | 2.37 (0.33) | 0.001 |
| C6 (C4:1-DC) | Hexanoylcarnitine (Fumarylcarnitine) | ETFDH | 4:159618737_G/A (rs1235904433) | stop gained | 2.1 × 10−13 (8.0 × 10−15) | 2.44 (0.33) | 0.001 |
| C8 | Octanoylcarnitine | ETFDH | 4:159618737_G/A (rs1235904433) | stop gained | 3.4 × 10−14 (6.5 × 10−18) | 2.51 (0.33) | 0.001 |
| C10:1 | Decenoylcarnitine | PPID (ETFDH) | 4:159631991_G/T (rs9410) | missense | 3.8 × 10−11 (2.7 × 10−11) | −0.14 (0.02) | 0.296 |
| C12 | Dodecanoylcarnitine | PPID (ETFDH) | 4:159631991_G/T (rs9410) | missense | 4.8 × 10−14 (5.2 × 10−14) | −0.17 (0.02) | 0.298 |
| C5-OH (C3-DC-M) | Hydroxyvalerylcarnitine (Methylmalonylcarnitine) | MCCC2 | 5:70952685_T/C (rs751970792) | stop lost | 1.9 × 10−12 (1.9 × 10−12) | 2.01 (0.28) | 0.001 |
| C16:1 | Hexadecenoylcarnitine | P4HA2; PDLIM4 (SLC22A5) | 5:131607402_T/C (rs10479000) | intron; intron | 2.3 × 10−10 (1.3 × 10−10) | −0.13 (0.02) | 0.489 |
| C2 | Acetylcarnitine | SLC22A5 | 5:131714129_G/A (rs386134194) | synonymous | 2.4 × 10−13 (3.2 × 10−13) | −1.38 (0.19) | 0.003 |
| C4 | Butyrylcarnitine | SLC22A5 | 5:131714129_G/A (rs386134194) | synonymous | 1.4 × 10−10 (1.3 × 10−12) | −1.22 (0.19) | 0.003 |
| C0 | Carnitine | SLC22A5 | 5:131714129_G/A (rs386134194) | synonymous | 5.6 × 10−12 (6.5 × 10−12) | −1.3 (0.19) | 0.003 |
| Asp | Aspartate | F12; GRK6 | 5:176836532_A/G (rs1801020) | 5UTR; intron | 2.5 × 10−9 (3.5 × 10−10) | 0.14 (0.02) | 0.235 |
| Taurine | Taurine | F12; GRK6 | 5:176836532_A/G (rs1801020) | 5UTR; intron | 5.9 × 10−16 (5.9 × 10−16) | 0.19 (0.02) | 0.235 |
| Sarcosine | Sarcosine | PEX6 (GNMT) | 6:42946943_G/A (rs9462859) | 5UTR | 7.8 × 10−13 (7.8 × 10−13) | −0.16 (0.02) | 0.478 |
| C3 | Propionylcarnitine | SLC22A1 | 6:160551204_G/C (rs683369) | missense | 7.7 × 10−12 (1.2 × 10−19) | 0.17 (0.03) | 0.196 |
| Serotonin | Serotonin | SLC22A1 | 6:160560880_CATG/C (rs72552763) | inframe insertion | 1.3 × 10−11 (1.2 × 10−11) | 0.19 (0.03) | 0.159 |
| Putrescine | Putrescine | AOC1 | 7:150553605_C/T (rs10156191) | missense | 2.3 × 10−1 (2.5 × 10−15) | 0.03 (0.02) | 0.236 |
| C10 | Decanoylcarnitine | COL27A1 | 9:116931401_C/T (rs145560419) | synonymous | 8.8 × 10−9 (3.2 × 10−10) | −2.68 (0.47) | 0.001 |
| C8 | Octanoylcarnitine | COL27A1 | 9:116931401_C/T (rs145560419) | synonymous | 4.7 × 10−9 (4.3 × 10−10) | −2.67 (0.46) | 0.001 |
| Sarcosine | Sarcosine | SARDH | 9:136598926_C/G (rs10993780) | intron | 5.3 × 10−44 (5.3 × 10−44) | −0.39 (0.03) | 0.171 |
| PC aa C36:0 | Phosphatidylcholine diacyl C36:0 | A1CF | 10:52603951_AT/A (-) | intron | 4.1 × 10−9 (5.4 × 10−9) | 2.23 (0.38) | 0.001 |
| Putrescine | Putrescine | JMJD1C | 10:65225899_A/AGGCGGC (rs3841602) | upstream | 1.7 × 10−19 (5.2 × 10−20) | 0.19 (0.02) | 0.477 |
| C16-OH | Hydroxyhexadecanoylcarnitine | PYROXD2 | 10:100148308_T/G (rs2147895) | intron | 8.9 × 10−19 (8.9 × 10−19) | −0.18 (0.02) | 0.336 |
| His | Histidine | PSMC3 | 11:47445720_G/A (rs186188306) | synonymous | 5.5 × 10−10 (4.5 × 10−9) | −2.34 (0.38) | 0.001 |
| lysoPC a C26:1 | lysoPhosphatidylcholine acyl C26:1 | TMEM258 (MYRF, FADS1, FADS2) | 11:61560081_G/A (rs174538) | 5UTR | 3.3 × 10−10 (3.3 × 10−10) | −0.14 (0.02) | 0.264 |
| Asn | Asparagine | ASRGL1 | 11:62105391_C/T (rs2513749) | 5UTR | 1.2 × 10−15 (7.8 × 10−19) | 0.24 (0.03) | 0.12 |
| Gln | Glutamine | GLS2 | 12:56866487_A/G (-) | missense | 8.2 × 10−13 (3.1 × 10−14) | −2.97 (0.41) | 0.001 |
| His | Histidine | TMPO | 12:98929093_A/G (rs867372792) | 3UTR | 8.2 × 10−15 (5.6 × 1013) | 1.72 (0.22) | 0.002 |
| His | Histidine | UHRF1BP1L (ACTR6) | 12:100492127_T/C (-) | splice region variant | 1.3 × 10−10 (2.6 × 10−9) | 1.32 (0.21) | 0.002 |
| Phe | Phenylalanine | PMCH | 12:102591269_G/T (rs200627654) | intron | 8.2 × 10−17 (1.4 × 10−14) | 1.3 (0.16) | 0.004 |
| His | Histidine | TCHP; GIT2 | 12:110385016_A/AG (-) | intron; intron | 4.2 × 10−12 (1.2 × 10−11) | 2.76 (0.4) | 0.001 |
| Asn | Asparagine | ASPG | 14:104576448_G/A (rs34362765) | intron | 8.8 × 10−105 (5.8 × 10−25) | −0.46 (0.02) | 0.358 |
| C10 | Decanoylcarnitine | ABCC1 | 16:16139714_T/C (rs35587) | synonymous | 4.5 × 10−8 (5.0 × 10−9) | −0.12 (0.02) | 0.326 |
| C12:1 | Dodecanoylcarnitine | ABCC1 | 16:16139714_T/C (rs35587) | synonymous | 7.2 × 10−10 (6.5 × 10−10) | −0.13 (0.02) | 0.326 |
| C12 | Dodecenoylcarnitine | ABCC1 | 16:16139714_T/C (rs35587) | synonymous | 1.2 × 10−9 (1.2 × 10−9) | −0.13 (0.02) | 0.326 |
| lysoPC a C20:3 | lysoPhosphatidylcholine acyl C20:3 | TM6SF2 | 19:19379549_C/T (rs58542926) | missense | 9.3 × 10−9 (5.0 × 10−9) | −0.25 (0.04) | 0.054 |
| PC aa C34:4 | Phosphatidylcholine diacyl C34:4 | TM6SF2 (SUGP1) | 19:19379549_C/T (rs58542926) | missense | 2.0 × 10−11 (4.1 × 10−12) | −0.29 (0.04) | 0.054 |
| Pro | Proline | PRODH | 22:18910479_C/T (rs13058335) | intron | 4.8 × 10−55 (4.5 × 10−31) | 0.68 (0.04) | 0.063 |
| Gene | Associated Metabolite(s) | Lead, LD, or Gene Variant 1 | Level | Description |
|---|---|---|---|---|
| A1CF | PC aa C36:0 | Lead | 2 | Apolipoprotein B (apo B) is a major component of low-density lipoproteins and in mammals exist in two isoforms: apoB-100 and apoB-48. The two isoforms are encoded by a single mRNA transcript. A1CF encodes an RNA binding protein that facilitates APOBEC1’s editing of APOB mRNA, introducing a premature stop codon that yields apoB-48, resulting in the truncated gene product known as apoB-48 [20]. ApoB-48 is produced by action of APOBEC-1 exclusively in the small intestine of humans and ApoB-48 can be found in chylomicrons synthetized in the small intestine. As expected, the present of not functional APOBEC-1 enzyme resulted in impaired circulating levels of triglycerides and cholesterol and we found that it also impacts on blood levels of several PCs, such as PC aa C36:0. |
| ABCC1 | C10, C12:1, C12 | Lead | 2 | This gene encodes for an ABC proteins that transport various molecules across extra-and intra-cellular membranes. |
| ACTR6 | His | LD | 3 | Actin Related Protein 6. The role of this gene is not fully understood as well as its association with histidine. |
| AOC1 | Putrescine | Lead | 1 | Amine oxidase copper containing 1 (AOC1) encodes a metal-binding membrane glycoprotein that oxidatively deaminates putrescine, histamine, and related compounds. |
| ASPG | Asn | Lead | 1 | Predicted to have lysophospholipase activity and mainly responsible to catalyze the conversion of asparagine to aspartate. |
| ASRGL1 | Asn | Lead | 1 | Encodes the l-asparaginase enzyme responsible for the catalysis of asparagine catabolism to aspartate. |
| CERS4 | SM C18:0 | Gene | 1 | This gene encodes for the protein Ceramide synthase 4, which catalyzes the formation of ceramides via sphinganine and acyl-CoA substrates, with high selectivity on long-chains. |
| COL27A1 | C10, C8 | Lead | 3 | The gene encodes a member of the fibrillar collagen family, involved in the cartilage calcification process and the transition of cartilage to bone. Mutations on this gene are known to cause Steel Syndrome. |
| ENPEP | Asp | Lead | 1 | ENPEP encodes for glutamyl aminopeptidase that regulates central hypertension through its calcium-modulated preference to cleave N-terminal acidic residues from peptides such as angiotensin II. This protein can upregulate blood pressure by cleaving the N-terminal aspartate from angiotensin II, and can regulate blood vessel formation and enhance tumorigenesis in some tissues. |
| ETFDH | C10, C6 (C4:1-DC), C8, C10:1, C12 | Lead/LD | 2 | This gene encodes for the Electron transfer flavoprotein (ETF) present in the mitochondria, which acts in the electron transfer for at least 9 flavins. Mutations on this gene (and other ETF genes such as ETFA and ETFB) are known to cause multiple acyl-CoA deficiency (MADD), also known as glutaric acidemia |
| FADS1 | PC ae C38:3, lysoPC a C26:1 | LD | 2 | Fatty acid desaturase enzymes regulate unsaturation of fatty acids through the introduction of double bonds into the fatty acyl chain. |
| FADS2 | PC ae C38:3, lysoPC a C26:1 | LD | 2 | Fatty acid desaturase enzymes regulate unsaturation of fatty acids through the introduction of double bonds into the fatty acyl chain. |
| F12; GRK6 | Asp, Taurine | Lead | 3 | The human coagulation factor XII (FXII) is involved in the intrinsic coagulation pathway. |
| GLS2 | Gln | Lead | 1 | The gene is responsible for encoding the glutaminase 2, an enzyme that catalyzes the conversion of glutamine to glutamate and ammonia, promoting mitochondrial respiration and ATP generation. |
| GNMT | Sarcosine | LD | 1 | Acts on the conversion of S-adenosyl-L-methionine (SAMe) and glycine to S-adenosyl-L-homocysteine and sarcosine. Defects in this gene are a cause of hypermethioninemia. |
| JMJD1C | Putrescine | Lead | 3 | Plays a central role in histone code and lysine demethylation. |
| LTA4H | His | LD | 3 | This gene encodes an enzyme used in the final step of the biosynthesis of leukotriene B4, a proinflammatory mediator. It is known to degrade proline-glycine-proline, biomarker for chronic obstructive pulmonary disease. |
| MCCC2 | C5-OH (C3-DC-M) | Lead | 2 | Catalyzes the conversion of 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA, playing an important role in the catabolism of leucine and isovaleric acid. Mutations in this gene are associated with 3-methylcrotonylglycinuria. |
| MTHFR | Ser | Lead | 2 | Responsible for the catalysis of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, involved in the remethylation of homocysteine to produce methionine and tetrahydrofolate, a substrate for serine production. |
| MYRF | PC ae C38:3, lysoPC a C26:1 | Lead, LD | 3 | Encodes an essential transcript factor that acts on the central nervous system myelination process. |
| P4HA2; PDLIM4 | C16:1 | Lead | 3 | P4HA2 gene encodes a component of prolyl 4-hydroxylase, a key enzyme in collagen synthesis. |
| PEX6 | Sarcosine | Lead | 3 | Encodes a member of the AAA family of ATPases, which plays a direct role in peroxisomal protein import and PTS1 (peroxisomal targeting signal 1, a C-terminal tripeptide of the sequence Ser-Lys-Leu) receptor activity. |
| PMCH | Phe | Lead | 3 | Responsible for the generation of multiple protein products including melanin-concentrating hormone (MCH), neuropeptide-glutamic acid-isoleucine (NEI), and neuropeptide-glycine-glutamic acid (NGE). Acts on behaviors such as hunger and arousal. |
| PPID | C10:1, C12 | Lead | 3 | Index variant associated with different carnitines and colocalized with decreased gene expression. PPID is a putatively novel gene. |
| PRODH | Pro | Lead | 1 | This protein catalysis the intermediate reaction of proline catabolism to glutamic acid and mutations on this gene are associated with hyperprolinemia type 1. |
| PSMC3 | His | Lead | 3 | Proteasome 26S Subunit, ATPase 3 (PSMC3) is a multicatalytic proteinase complex. |
| PYROXD2 | C16-OH | Lead | 3 | Predicted oxidoreductase that may play in mitochondrial organization. |
| SARDH | Sarcosine | Lead | 1 | This gene encodes for the sarcosine dehydrogenase enzyme that acts on the conversion of sarcosine to glycine. Mutations in this gene are the cause for sarcosinemia. |
| SLC22A1 | C3, Serotonin | Lead | 1 | An organic cation transporter with polyspecificity, such as for histamine, epinephrine, adrenaline, noradrenaline, dopamine, spermine and spermidine, among others. |
| SLC22A5 | C0, C2, C4, C16:1 | Lead/LD/gene | 1 | An organic cation transporter with high affinity for carnitine. Mutations in this gene are the cause of systemic primary carnitine deficiency. |
| SUGP1 | PC aa C34:4 | LD | 3 | Acts in pre-mRNA splicing. |
| TCHP; GIT2 | His | Lead | 3 | Trichoplein keratin filament binding (TCHP) encodes for a protein with unknown function. |
| TDO2 | Trp | Gene | 1 | This enzyme catalyzes the first and rate-limiting step in the conversion of tryptophan into kynurenine. |
| TM6SF2 | lysoPC a C20:3, PC aa C34:4 | Lead | 2 | Regulator of liver fat metabolism this gene influences triglyceride secretion and hepatic lipid droplet content. It is associated with fatty liver disease and non-alcoholic fatty liver disease. |
| TMEM258 | lysoPC a C26:1, PC ae C38:3 | Lead | 3 | Transmembrane Protein 258 (TMEM258) is a component of the oligosaccharyltransferase complex controlling ER stress and intestinal inflammation. |
| TMPO | His | Lead | 3 | This gene encodes several proteins containing a LEM domain through an alternative splicing mechanism. These proteins are involved in gene expression, chromatin organization, replication and cell cycle control. |
| UHRF1BP1L | His | Lead | 3 | UHRF1 Binding Protein 1 Like (URHF1BP1L) has analogy with ubiquitin-like containing PHD and RING finger domains. |
| Trait ID | Trait Name | Gene | Mask 1 | p-Value (Conditional) | Number of Variants | Cumulative Allele Count | Number of Variants Needed to Reach Significance |
|---|---|---|---|---|---|---|---|
| Trp | Tryptophan | TDO2 | HMI | 8.9 × 10−9 (1.7 × 10−8) | 6 | 45 | 3 |
| SM C18:0 | Sphingomyeline C18:0 | CERS4 | HMI | 6.1 × 10−10 (2.7 × 10−8) | 10 | 82 | 3 |
| C0 | Carnitine | SLC22A5 | HMI | 2.9 × 10−10 (4.0 × 10−10) | 6 | 37 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
König, E.; Rainer, J.; Hernandes, V.V.; Paglia, G.; Del Greco M., F.; Bottigliengo, D.; Yin, X.; Chan, L.S.; Teumer, A.; Pramstaller, P.P.; et al. Whole Exome Sequencing Enhanced Imputation Identifies 85 Metabolite Associations in the Alpine CHRIS Cohort. Metabolites 2022, 12, 604. https://doi.org/10.3390/metabo12070604
König E, Rainer J, Hernandes VV, Paglia G, Del Greco M. F, Bottigliengo D, Yin X, Chan LS, Teumer A, Pramstaller PP, et al. Whole Exome Sequencing Enhanced Imputation Identifies 85 Metabolite Associations in the Alpine CHRIS Cohort. Metabolites. 2022; 12(7):604. https://doi.org/10.3390/metabo12070604
Chicago/Turabian StyleKönig, Eva, Johannes Rainer, Vinicius Verri Hernandes, Giuseppe Paglia, Fabiola Del Greco M., Daniele Bottigliengo, Xianyong Yin, Lap Sum Chan, Alexander Teumer, Peter P. Pramstaller, and et al. 2022. "Whole Exome Sequencing Enhanced Imputation Identifies 85 Metabolite Associations in the Alpine CHRIS Cohort" Metabolites 12, no. 7: 604. https://doi.org/10.3390/metabo12070604
APA StyleKönig, E., Rainer, J., Hernandes, V. V., Paglia, G., Del Greco M., F., Bottigliengo, D., Yin, X., Chan, L. S., Teumer, A., Pramstaller, P. P., Locke, A. E., & Fuchsberger, C. (2022). Whole Exome Sequencing Enhanced Imputation Identifies 85 Metabolite Associations in the Alpine CHRIS Cohort. Metabolites, 12(7), 604. https://doi.org/10.3390/metabo12070604