
High-Density Lipoprotein Alterations in Type 2 Diabetes and Obesity

 , ,
, ,
Abstract
:
1. Introduction
2. HDL Size and Composition
2.1. Particle Size
2.2. Lipidome
2.3. Proteome
2.4. Glycation and Oxidation
2.5. Carbamylation
3. HDL/apoA-I Kinetics
4. HDL Functions
4.1. Reverse Cholesterol Transport
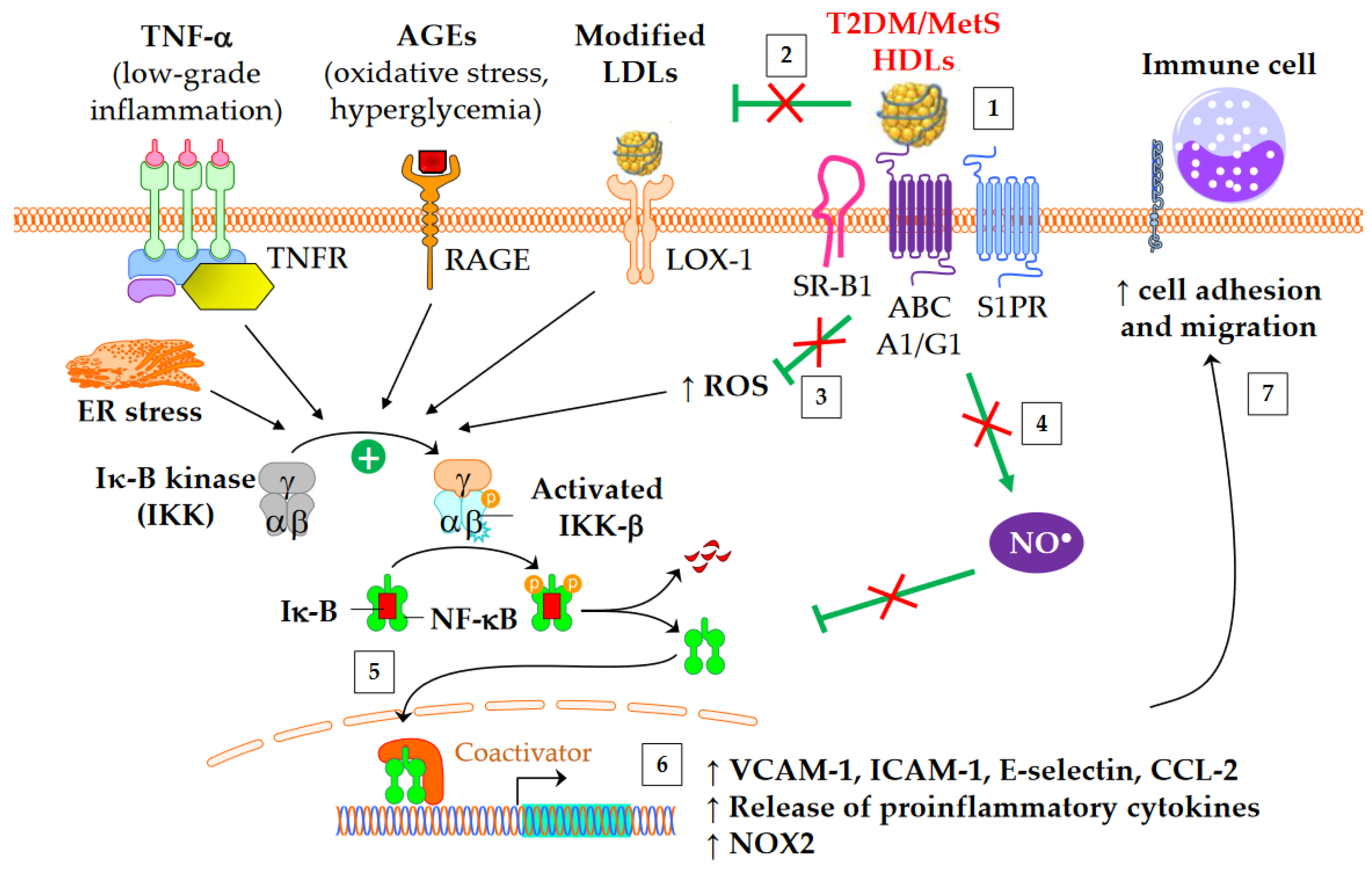
4.2. Anti-Inflammatory Properties
4.3. Antioxidative Properties
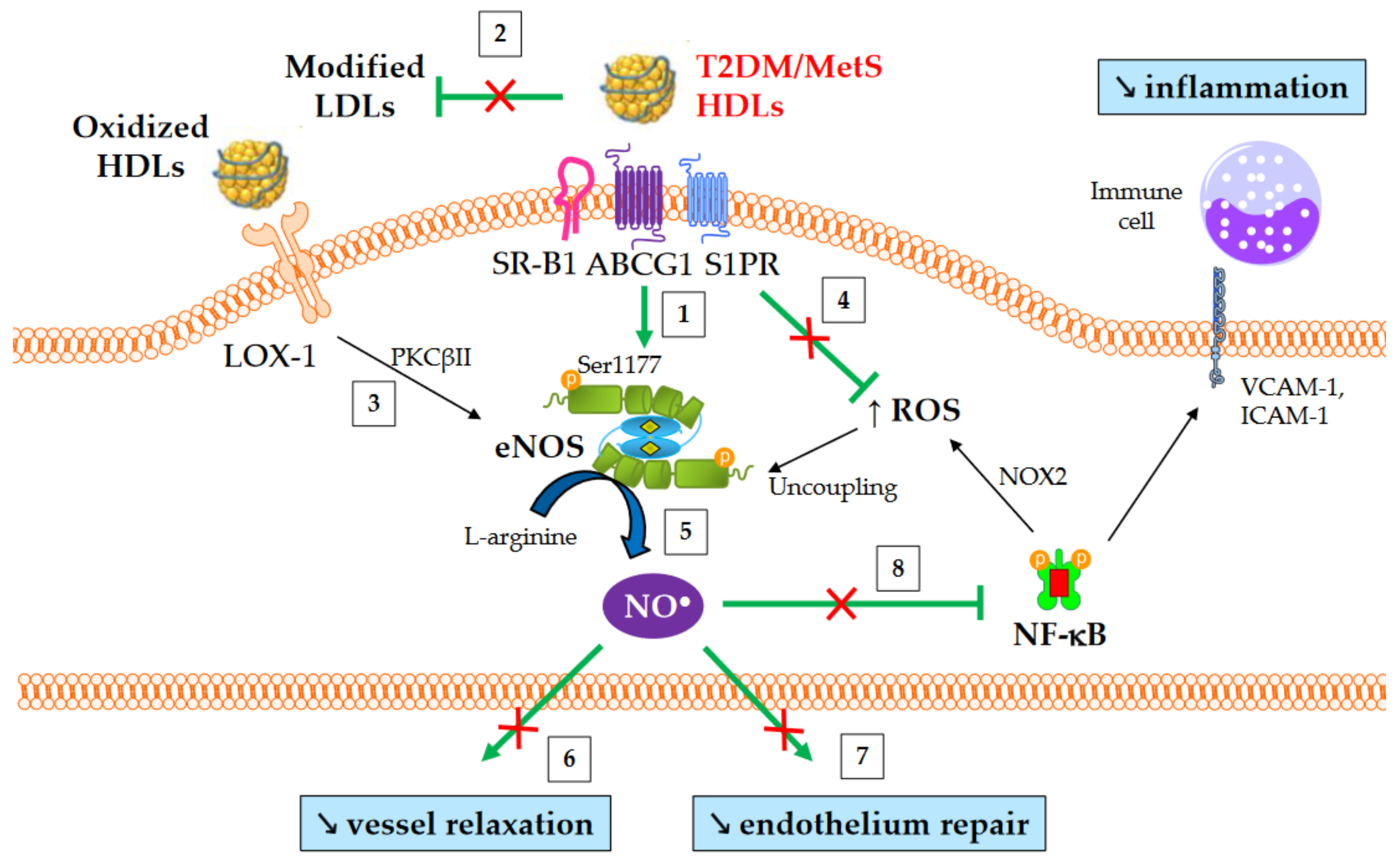
4.4. Nitric Oxide Production
4.5. Antiapoptotic Properties and Endothelium Repair
5. Antidiabetic Properties
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IDF Diabetes Atlas. Available online: https://diabetesatlas.org/ (accessed on 2 February 2023).
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-Mapping Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Islet-Specific Epigenome Maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Dorcely, B.; Katz, K.; Jagannathan, R.; Chiang, S.S.; Oluwadare, B.; Goldberg, I.J.; Bergman, M. Novel Biomarkers for Prediabetes, Diabetes, and Associated Complications. Diabetes Metab. Syndr. Obes. 2017, 10, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from Coronary Heart Disease in Subjects with Type 2 Diabetes and in Nondiabetic Subjects with and without Prior Myocardial Infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef] [PubMed]
- Guembe, M.J.; Fernandez-Lazaro, C.I.; Sayon-Orea, C.; Toledo, E.; Moreno-Iribas, C.; Cosials, J.B.; Reyero, J.B.; Martínez, J.D.; Diego, P.G.; Uche, A.M.G.; et al. Risk for Cardiovascular Disease Associated with Metabolic Syndrome and Its Components: A 13-Year Prospective Study in the RIVANA Cohort. Cardiovasc. Diabetol. 2020, 19, 195. [Google Scholar] [CrossRef]
- Hirano, T. Pathophysiology of Diabetic Dyslipidemia. J. Atheroscler. Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef]
- Bonilha, I.; Hajduch, E.; Luchiari, B.; Nadruz, W.; Le Goff, W.; Sposito, A.C. The Reciprocal Relationship between LDL Metabolism and Type 2 Diabetes Mellitus. Metabolites 2021, 11, 807. [Google Scholar] [CrossRef]
- Wilson, P.W.; Abbott, R.D.; Castelli, W.P. High Density Lipoprotein Cholesterol and Mortality. The Framingham Heart Study. Arteriosclerosis 1988, 8, 737–741. [Google Scholar] [CrossRef]
- Ko, D.T.; Alter, D.A.; Guo, H.; Koh, M.; Lau, G.; Austin, P.C.; Booth, G.L.; Hogg, W.; Jackevicius, C.A.; Lee, D.S.; et al. High-Density Lipoprotein Cholesterol and Cause-Specific Mortality in Individuals without Previous Cardiovascular Conditions: The CANHEART Study. J. Am. Coll. Cardiol. 2016, 68, 2073–2083. [Google Scholar] [CrossRef]
- Von Eckardstein, A.; Nordestgaard, B.G.; Remaley, A.T.; Catapano, A.L. High-Density Lipoprotein Revisited: Biological Functions and Clinical Relevance. Eur. Heart J. 2022, ahead of print, ehac605. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme High High-Density Lipoprotein Cholesterol Is Paradoxically Associated with High Mortality in Men and Women: Two Prospective Cohort Studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef]
- Liu, C.; Dhindsa, D.; Almuwaqqat, Z.; Sun, Y.V.; Quyyumi, A.A. Very High High-Density Lipoprotein Cholesterol Levels and Cardiovascular Mortality. Am. J. Cardiol. 2022, 167, 43–53. [Google Scholar] [CrossRef]
- Liu, C.; Dhindsa, D.; Almuwaqqat, Z.; Ko, Y.-A.; Mehta, A.; Alkhoder, A.A.; Alras, Z.; Desai, S.R.; Patel, K.J.; Hooda, A.; et al. Association Between High-Density Lipoprotein Cholesterol Levels and Adverse Cardiovascular Outcomes in High-Risk Populations. JAMA Cardiol. 2022, 7, 672–680. [Google Scholar] [CrossRef]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare Variant in Scavenger Receptor BI Raises HDL Cholesterol and Increases Risk of Coronary Heart Disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef]
- Haase, C.L.; Tybjærg-Hansen, A.; Qayyum, A.A.; Schou, J.; Nordestgaard, B.G.; Frikke-Schmidt, R. LCAT, HDL Cholesterol and Ischemic Cardiovascular Disease: A Mendelian Randomization Study of HDL Cholesterol in 54,500 Individuals. J. Clin. Endocrinol. Metab. 2012, 97, E248–E256. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.A.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjaerg-Hansen, A. Association of Loss-of-Function Mutations in the ABCA1 Gene with High-Density Lipoprotein Cholesterol Levels and Risk of Ischemic Heart Disease. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef]
- Apro, J.; Tietge, U.J.F.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated from Interstitial Fluid in Type 2 Diabetes Mellitus-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef]
- Ståhlman, M.; Fagerberg, B.; Adiels, M.; Ekroos, K.; Chapman, J.M.; Kontush, A.; Borén, J. Dyslipidemia, but Not Hyperglycemia and Insulin Resistance, Is Associated with Marked Alterations in the HDL Lipidome in Type 2 Diabetic Subjects in the DIWA Cohort: Impact on Small HDL Particles. Biochim. Biophys. Acta 2013, 1831, 1609–1617. [Google Scholar] [CrossRef]
- Mora, S.; Otvos, J.D.; Rosenson, R.S.; Pradhan, A.; Buring, J.E.; Ridker, P.M. Lipoprotein Particle Size and Concentration by Nuclear Magnetic Resonance and Incident Type 2 Diabetes in Women. Diabetes 2010, 59, 1153–1160. [Google Scholar] [CrossRef]
- Sokooti, S.; Flores-Guerrero, J.L.; Kieneker, L.M.; Heerspink, H.J.L.; Connelly, M.A.; Bakker, S.J.L.; Dullaart, R.P.F. HDL Particle Subspecies and Their Association with Incident Type 2 Diabetes: The PREVEND Study. J. Clin. Endocrinol. Metab. 2021, 106, 1761–1772. [Google Scholar] [CrossRef]
- Cardner, M.; Yalcinkaya, M.; Goetze, S.; Luca, E.; Balaz, M.; Hunjadi, M.; Hartung, J.; Shemet, A.; Kränkel, N.; Radosavljevic, S.; et al. Structure-Function Relationships of HDL in Diabetes and Coronary Heart Disease. JCI Insight 2020, 5, e131491. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Heink, A.; Sexmith, H.; Dolan, L.M.; Gordon, S.M.; Otvos, J.D.; Melchior, J.T.; Elder, D.A.; Khoury, J.; Geh, E.; et al. Obesity Is Associated with an Altered HDL Subspecies Profile among Adolescents with Metabolic Disease. J. Lipid Res. 2017, 58, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Kwon, S.; Zheng, D.; Shaughnessy, S.; Wallace, P.; Hutto, A.; Pugh, K.; Jenkins, A.J.; Klein, R.L.; Liao, Y. Effects of Insulin Resistance and Type 2 Diabetes on Lipoprotein Subclass Particle Size and Concentration Determined by Nuclear Magnetic Resonance. Diabetes 2003, 52, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.; Williams, K.; Hanley, A.J.G.; Otvos, J.D.; Goff, D.C.; Wagenknecht, L.E.; Haffner, S.M. Nuclear Magnetic Resonance Lipoprotein Abnormalities in Prediabetic Subjects in the Insulin Resistance Atherosclerosis Study. Circulation 2005, 111, 3465–3472. [Google Scholar] [CrossRef]
- Tabara, Y.; Arai, H.; Hirao, Y.; Takahashi, Y.; Setoh, K.; Kawaguchi, T.; Kosugi, S.; Ito, Y.; Nakayama, T.; Matsuda, F. Different Inverse Association of Large High-Density Lipoprotein Subclasses with Exacerbation of Insulin Resistance and Incidence of Type 2 Diabetes: The Nagahama Study. Diabetes Res. Clin. Pract. 2017, 127, 123–131. [Google Scholar] [CrossRef]
- Low, H.; Hoang, A.; Forbes, J.; Thomas, M.; Lyons, J.G.; Nestel, P.; Bach, L.A.; Sviridov, D. Advanced Glycation End-Products (AGEs) and Functionality of Reverse Cholesterol Transport in Patients with Type 2 Diabetes and in Mouse Models. Diabetologia 2012, 55, 2513–2521. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Groen, A.K.; Dallinga-Thie, G.M.; de Vries, R.; Sluiter, W.J.; van Tol, A. Fibroblast Cholesterol Efflux to Plasma from Metabolic Syndrome Subjects Is Not Defective despite Low High-Density Lipoprotein Cholesterol. Eur. J. Endocrinol. 2008, 158, 53–60. [Google Scholar] [CrossRef]
- Guérin, M.; Le Goff, W.; Lassel, T.S.; Van Tol, A.; Steiner, G.; Chapman, M.J. Proatherogenic Role of Elevated CE Transfer from HDL to VLDL1 and Dense LDL in Type 2 Diabetes. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 282–288. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Pagano, S.; Perton, F.G.; Vuilleumier, N. Antibodies Against the C-Terminus of ApoA-1 Are Inversely Associated with Cholesterol Efflux Capacity and HDL Metabolism in Subjects with and without Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 732. [Google Scholar] [CrossRef]
- Bouillet, B.; Gautier, T.; Blache, D.; Pais de Barros, J.-P.; Duvillard, L.; Petit, J.-M.; Lagrost, L.; Vergès, B. Glycation of Apolipoprotein C1 Impairs Its CETP Inhibitory Property: Pathophysiological Relevance in Patients with Type 1 and Type 2 Diabetes. Diabetes Care 2014, 37, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Rouland, A.; Masson, D.; Lagrost, L.; Vergès, B.; Gautier, T.; Bouillet, B. Role of Apolipoprotein C1 in Lipoprotein Metabolism, Atherosclerosis and Diabetes: A Systematic Review. Cardiovasc. Diabetol. 2022, 21, 272. [Google Scholar] [CrossRef]
- Shiu, S.W.M.; Tan, K.C.B.; Huang, Y.; Wong, Y. Type 2 Diabetes Mellitus and Endothelial Lipase. Atherosclerosis 2008, 198, 441–447. [Google Scholar] [CrossRef]
- Shiu, S.W.; Zhou, H.; Wong, Y.; Tan, K.C. Endothelial Lipase and Reverse Cholesterol Transport in Type 2 Diabetes Mellitus. J. Diabetes Investig. 2010, 1, 111–116. [Google Scholar] [CrossRef]
- Badellino, K.O.; Wolfe, M.L.; Reilly, M.P.; Rader, D.J. Endothelial Lipase Concentrations Are Increased in Metabolic Syndrome and Associated with Coronary Atherosclerosis. PLoS Med. 2006, 3, e22. [Google Scholar] [CrossRef]
- Shiu, S.W.; Wong, Y.; Tan, K.C. Pre-Β1 HDL in Type 2 Diabetes Mellitus. Atherosclerosis 2017, 263, 24–28. [Google Scholar] [CrossRef]
- Zhou, H.; Tan, K.C.B.; Shiu, S.W.M.; Wong, Y. Cellular Cholesterol Efflux to Serum Is Impaired in Diabetic Nephropathy. Diabetes Metab. Res. Rev. 2008, 24, 617–623. [Google Scholar] [CrossRef]
- Hirayama, S.; Miida, T.; Miyazaki, O.; Aizawa, Y. Preβ1-HDL Concentration Is a Predictor of Carotid Atherosclerosis in Type 2 Diabetic Patients. Diabetes Care 2007, 30, 1289–1291. [Google Scholar] [CrossRef]
- Vergès, B.; Florentin, E.; Baillot-Rudoni, S.; Petit, J.-M.; Brindisi, M.C.; Pais de Barros, J.-P.; Lagrost, L.; Gambert, P.; Duvillard, L. Rosuvastatin 20 Mg Restores Normal HDL-ApoA-I Kinetics in Type 2 Diabetes. J. Lipid Res. 2009, 50, 1209–1215. [Google Scholar] [CrossRef]
- Kostara, C.E.; Karakitsou, K.S.; Florentin, M.; Bairaktari, E.T.; Tsimihodimos, V. Progressive, Qualitative, and Quantitative Alterations in HDL Lipidome from Healthy Subjects to Patients with Prediabetes and Type 2 Diabetes. Metabolites 2022, 12, 683. [Google Scholar] [CrossRef]
- Denimal, D.; Benanaya, S.; Monier, S.; Simoneau, I.; Pais de Barros, J.-P.; Le Goff, W.; Bouillet, B.; Vergès, B.; Duvillard, L. Normal HDL Cholesterol Efflux and Anti-Inflammatory Capacities in Type 2 Diabetes despite Lipidomic Abnormalities. J. Clin. Endocrinol. Metab. 2022, 107, e3816–e3823. [Google Scholar] [CrossRef]
- Mocciaro, G.; D’Amore, S.; Jenkins, B.; Kay, R.; Murgia, A.; Herrera-Marcos, L.V.; Neun, S.; Sowton, A.P.; Hall, Z.; Palma-Duran, S.A.; et al. Lipidomic Approaches to Study HDL Metabolism in Patients with Central Obesity Diagnosed with Metabolic Syndrome. IJMS 2022, 23, 6786. [Google Scholar] [CrossRef] [PubMed]
- Duvillard, L.; Florentin, E.; Pont, F.; Petit, J.-M.; Baillot-Rudoni, S.; Penfornis, A.; Vergès, B. Chronic Hyperinsulinemia Does Not Increase the Production Rate of High-Density Lipoprotein Apolipoprotein AI: Evidence from a Kinetic Study in Patients with Insulinoma. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2460–2465. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B.; Petit, J.M.; Duvillard, L.; Dautin, G.; Florentin, E.; Galland, F.; Gambert, P. Adiponectin Is an Important Determinant of ApoA-I Catabolism. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K.; Bonnet, D.J.; Rye, K.A. The Conformation of Apolipoprotein A-I in High-Density Lipoproteins Is Influenced by Core Lipid Composition and Particle Size: A Surface Plasmon Resonance Study. Biochemistry 2000, 39, 5712–5721. [Google Scholar] [CrossRef]
- Denimal, D.; Nguyen, A.; Pais de Barros, J.-P.; Bouillet, B.; Petit, J.-M.; Vergès, B.; Duvillard, L. Major Changes in the Sphingophospholipidome of HDL in Non-Diabetic Patients with Metabolic Syndrome. Atherosclerosis 2016, 246, 106–114. [Google Scholar] [CrossRef]
- Nelson, T.L.; Kamineni, A.; Psaty, B.; Cushman, M.; Jenny, N.S.; Hokanson, J.; Furberg, C.; Mukamal, K.J. Lipoprotein-Associated Phospholipase A(2) and Future Risk of Subclinical Disease and Cardiovascular Events in Individuals with Type 2 Diabetes: The Cardiovascular Health Study. Diabetologia 2011, 54, 329–333. [Google Scholar] [CrossRef]
- Jackisch, L.; Kumsaiyai, W.; Moore, J.D.; Al-Daghri, N.; Kyrou, I.; Barber, T.M.; Randeva, H.; Kumar, S.; Tripathi, G.; McTernan, P.G. Differential Expression of Lp-PLA2 in Obesity and Type 2 Diabetes and the Influence of Lipids. Diabetologia 2018, 61, 1155–1166. [Google Scholar] [CrossRef]
- Kappelle, P.J.W.H.; de Boer, J.F.; Perton, F.G.; Annema, W.; de Vries, R.; Dullaart, R.P.F.; Tietge, U.J.F. Increased LCAT Activity and Hyperglycaemia Decrease the Antioxidative Functionality of HDL. Eur. J. Clin. Investig. 2012, 42, 487–495. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Perton, F.; Sluiter, W.J.; de Vries, R.; van Tol, A. Plasma Lecithin: Cholesterol Acyltransferase Activity Is Elevated in Metabolic Syndrome and Is an Independent Marker of Increased Carotid Artery Intima Media Thickness. J. Clin. Endocrinol. Metab. 2008, 93, 4860–4866. [Google Scholar] [CrossRef]
- Taradeh, M.; Dahik, V.; Lhomme, M.; Galier, S.; Hardy, L.; Frisdal, E.; Durand, H.; Kontush, A.; Bruckert, E.; Giral, P.; et al. Enrichment of High-Density Lipoproteins with Phosphatidylethanolamine (36:5) Impairs Their Protective Biological Activities and Is Associated with Atherosclerosis in Women. Atherosclerosis 2022, 355, 72. [Google Scholar] [CrossRef]
- Sutter, I.; Velagapudi, S.; Othman, A.; Riwanto, M.; Manz, J.; Rohrer, L.; Rentsch, K.; Hornemann, T.; Landmesser, U.; von Eckardstein, A. Plasmalogens of High-Density Lipoproteins (HDL) Are Associated with Coronary Artery Disease and Anti-Apoptotic Activity of HDL. Atherosclerosis 2015, 241, 539–546. [Google Scholar] [CrossRef]
- Kurano, M.; Tsukamoto, K.; Shimizu, T.; Kassai, H.; Nakao, K.; Aiba, A.; Hara, M.; Yatomi, Y. Protection against Insulin Resistance by Apolipoprotein M/Sphingosine-1-Phosphate. Diabetes 2020, 69, 867–881. [Google Scholar] [CrossRef]
- Vaisar, T.; Couzens, E.; Hwang, A.; Russell, M.; Barlow, C.E.; DeFina, L.F.; Hoofnagle, A.N.; Kim, F. Type 2 Diabetes Is Associated with Loss of HDL Endothelium Protective Functions. PLoS ONE 2018, 13, e0192616. [Google Scholar] [CrossRef]
- Plomgaard, P.; Dullaart, R.P.F.; de Vries, R.; Groen, A.K.; Dahlbäck, B.; Nielsen, L.B. Apolipoprotein M Predicts Pre-Beta-HDL Formation: Studies in Type 2 Diabetic and Nondiabetic Subjects. J. Intern. Med. 2009, 266, 258–267. [Google Scholar] [CrossRef]
- Memon, A.A.; Bennet, L.; Zöller, B.; Wang, X.; Palmér, K.; Dahlbäck, B.; Sundquist, J.; Sundquist, K. The Association between Apolipoprotein M and Insulin Resistance Varies with Country of Birth. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1174–1180. [Google Scholar] [CrossRef]
- Zhang, P.; Gao, J.; Pu, C.; Feng, G.; Wang, L.; Huang, L.; Tao, Q.; Zhang, Y. Effects of Hyperlipidaemia on Plasma Apolipoprotein M Levels in Patients with Type 2 Diabetes Mellitus: An Independent Case-Control Study. Lipids Health Dis. 2016, 15, 158. [Google Scholar] [CrossRef]
- Mughal, S.A.; Park, R.; Nowak, N.; Gloyn, A.L.; Karpe, F.; Matile, H.; Malecki, M.T.; McCarthy, M.I.; Stoffel, M.; Owen, K.R. Apolipoprotein M Can Discriminate HNF 1A-MODY from Type 1 Diabetes. Diabet. Med. 2013, 30, 246–250. [Google Scholar] [CrossRef]
- Tong, X.; Peng, H.; Liu, D.; Ji, L.; Niu, C.; Ren, J.; Pan, B.; Hu, J.; Zheng, L.; Huang, Y. High-Density Lipoprotein of Patients with Type 2 Diabetes Mellitus Upregulates Cyclooxgenase-2 Expression and Prostacyclin I-2 Release in Endothelial Cells: Relationship with HDL-Associated Sphingosine-1-Phosphate. Cardiovasc. Diabetol. 2013, 12, 27. [Google Scholar] [CrossRef]
- Zhao, D.; Yang, L.-Y.; Wang, X.-H.; Yuan, S.-S.; Yu, C.-G.; Wang, Z.-W.; Lang, J.-N.; Feng, Y.-M. Different Relationship between ANGPTL3 and HDL Components in Female Non-Diabetic Subjects and Type-2 Diabetic Patients. Cardiovasc. Diabetol. 2016, 15, 132. [Google Scholar] [CrossRef]
- Denimal, D.; Monier, S.; Brindisi, M.-C.; Petit, J.-M.; Bouillet, B.; Nguyen, A.; Demizieux, L.; Simoneau, I.; Pais de Barros, J.-P.; Vergès, B.; et al. Impairment of the Ability of HDL from Patients with Metabolic Syndrome but without Diabetes Mellitus to Activate ENOS: Correction by S1P Enrichment. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, M.C.; Shanmugarajah, N.; Lee, S.X.; Kraakman, M.J.; Westerterp, M.; Kitamoto, T.; Harris, M.; Cook, J.R.; Gusarova, G.A.; Zhong, K.; et al. Hepatic FoxOs Link Insulin Signaling with Plasma Lipoprotein Metabolism through an Apolipoprotein M/Sphingosine-1-Phosphate Pathway. J. Clin. Investig. 2022, 132, e146219. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.; Lehmann, I.; Gräler, M.; Bröcker-Preuss, M.; Erbel, R.; Heusch, G.; Levkau, B. HDL-Bound Sphingosine 1-Phosphate (S1P) Predicts the Severity of Coronary Artery Atherosclerosis. Cell. Physiol. Biochem. 2014, 34, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, B.; Luo, G.; Nilsson-Ehle, P.; Xu, N. Hyperglycemia Down-Regulates Apolipoprotein M Expression in Vivo and in Vitro. Biochim. Biophys. Acta 2007, 1771, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kurano, M.; Nanya, M.; Shimizu, T.; Ohkawa, R.; Tozuka, M.; Yatomi, Y. Glycation of HDL Polymerizes Apolipoprotein M and Attenuates Its Capacity to Bind to Sphingosine 1-Phosphate. J. Atheroscler. Thromb. 2021, 28, 730–741. [Google Scholar] [CrossRef]
- HDL Proteome Watch Page. Available online: https://homepages.uc.edu/~davidswm/HDLproteome.html (accessed on 25 November 2022).
- Holzer, M.; Ljubojevic-Holzer, S.; Souza Junior, D.R.; Stadler, J.T.; Rani, A.; Scharnagl, H.; Ronsein, G.E.; Marsche, G. HDL Isolated by Immunoaffinity, Ultracentrifugation, or Precipitation Is Compositionally and Functionally Distinct. J. Lipid Res. 2022, 63, 100307. [Google Scholar] [CrossRef]
- Tsun, J.G.S.; Shiu, S.W.M.; Wong, Y.; Yung, S.; Chan, T.M.; Tan, K.C.B. Impact of Serum Amyloid A on Cellular Cholesterol Efflux to Serum in Type 2 Diabetes Mellitus. Atherosclerosis 2013, 231, 405–410. [Google Scholar] [CrossRef]
- Annema, W.; Nijstad, N.; Tölle, M.; de Boer, J.F.; Buijs, R.V.C.; Heeringa, P.; van der Giet, M.; Tietge, U.J.F. Myeloperoxidase and Serum Amyloid A Contribute to Impaired in Vivo Reverse Cholesterol Transport during the Acute Phase Response but Not Group IIA Secretory Phospholipase A(2). J. Lipid Res. 2010, 51, 743–754. [Google Scholar] [CrossRef]
- Kronenberg, F.; Stühlinger, M.; Trenkwalder, E.; Geethanjali, F.S.; Pachinger, O.; von Eckardstein, A.; Dieplinger, H. Low Apolipoprotein A-IV Plasma Concentrations in Men with Coronary Artery Disease. J. Am. Coll. Cardiol. 2000, 36, 751–757. [Google Scholar] [CrossRef]
- Qu, J.; Ko, C.-W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Liu, D.; Ji, L.; Zhao, M.; Wang, Y.; Guo, Y.; Li, L.; Zhang, D.; Xu, L.; Pan, B.; Su, J.; et al. Lysine Glycation of Apolipoprotein A-I Impairs Its Anti-Inflammatory Function in Type 2 Diabetes Mellitus. J. Mol. Cell. Cardiol. 2018, 122, 47–57. [Google Scholar] [CrossRef]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.; Thornalley, P.J.; Rabbani, N. Arginine-Directed Glycation and Decreased HDL Plasma Concentration and Functionality. Nutr. Diabetes 2014, 4, e134. [Google Scholar] [CrossRef]
- Bacchetti, T.; Masciangelo, S.; Armeni, T.; Bicchiega, V.; Ferretti, G. Glycation of Human High Density Lipoprotein by Methylglyoxal: Effect on HDL-Paraoxonase Activity. Metabolism 2014, 63, 307–311. [Google Scholar] [CrossRef]
- Hoang, A.; Murphy, A.J.; Coughlan, M.T.; Thomas, M.C.; Forbes, J.M.; O’Brien, R.; Cooper, M.E.; Chin-Dusting, J.P.F.; Sviridov, D. Advanced Glycation of Apolipoprotein A-I Impairs Its Anti-Atherogenic Properties. Diabetologia 2007, 50, 1770–1779. [Google Scholar] [CrossRef]
- Aroor, A.R.; DeMarco, V.G. Oxidative Stress and Obesity: The Chicken or the Egg? Diabetes 2014, 63, 2216–2218. [Google Scholar] [CrossRef]
- Marin, M.T.; Dasari, P.S.; Tryggestad, J.B.; Aston, C.E.; Teague, A.M.; Short, K.R. Oxidized HDL and LDL in Adolescents with Type 2 Diabetes Compared to Normal Weight and Obese Peers. J. Diabetes Complicat. 2015, 29, 679–685. [Google Scholar] [CrossRef]
- Sartore, G.; Seraglia, R.; Burlina, S.; Bolis, A.; Marin, R.; Manzato, E.; Ragazzi, E.; Traldi, P.; Lapolla, A. High-Density Lipoprotein Oxidation in Type 2 Diabetic Patients and Young Patients with Premature Myocardial Infarction. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 418–425. [Google Scholar] [CrossRef]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-Vasoprotective Effects of High-Density Lipoprotein Are Impaired in Patients with Type 2 Diabetes Mellitus but Are Improved after Extended-Release Niacin Therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef]
- Feng, J.; Wang, Y.; Li, W.; Zhao, Y.; Liu, Y.; Yao, X.; Liu, S.; Yu, P.; Li, R. High Levels of Oxidized Fatty Acids in HDL Impair the Antioxidant Function of HDL in Patients with Diabetes. Front. Endocrinol. 2022, 13, 993193. [Google Scholar] [CrossRef]
- Pérez, L.; Vallejos, A.; Echeverria, C.; Varela, D.; Cabello-Verrugio, C.; Simon, F. OxHDL Controls LOX-1 Expression and Plasma Membrane Localization through a Mechanism Dependent on NOX/ROS/NF-ΚB Pathway on Endothelial Cells. Lab. Investig. 2019, 99, 421–437. [Google Scholar] [CrossRef]
- Assinger, A.; Koller, F.; Schmid, W.; Zellner, M.; Babeluk, R.; Koller, E.; Volf, I. Specific Binding of Hypochlorite-Oxidized HDL to Platelet CD36 Triggers Proinflammatory and Procoagulant Effects. Atherosclerosis 2010, 212, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, J.J.; Meuwese, M.C.; van Miert, J.N.I.; Kastelein, A.; Tijssen, J.G.P.; Piek, J.J.; Trip, M.D. Diabetes Mellitus Type 2 Is Associated with Higher Levels of Myeloperoxidase. Med. Sci. Monit. 2008, 14, CR406–CR410. [Google Scholar] [PubMed]
- Shiu, S.W.M.; Xiao, S.-M.; Wong, Y.; Chow, W.-S.; Lam, K.S.L.; Tan, K.C.B. Carbamylation of LDL and Its Relationship with Myeloperoxidase in Type 2 Diabetes Mellitus. Clin. Sci. 2013, 126, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.B.; Cheung, C.-L.; Lee, A.C.H.; Lam, J.K.Y.; Wong, Y.; Shiu, S.W.M. Carbamylated Lipoproteins and Progression of Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 359–366. [Google Scholar] [CrossRef]
- Chen, Z.; Ding, S.; Wang, Y.P.; Chen, L.; Mao, J.Y.; Yang, Y.; Sun, J.T.; Yang, K. Association of Carbamylated High-Density Lipoprotein with Coronary Artery Disease in Type 2 Diabetes Mellitus: Carbamylated High-Density Lipoprotein of Patients Promotes Monocyte Adhesion. J. Transl. Med. 2020, 18, 460. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein Carbamylation Links Inflammation, Smoking, Uremia and Atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Lui, D.T.W.; Cheung, C.-L.; Lee, A.C.H.; Wong, Y.; Shiu, S.W.M.; Tan, K.C.B. Carbamylated HDL and Mortality Outcomes in Type 2 Diabetes. Diabetes Care 2021, 44, 804–809. [Google Scholar] [CrossRef]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein Carbamylation Renders High-Density Lipoprotein Dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef]
- Denimal, D.; Monier, S.; Simoneau, I.; Duvillard, L.; Vergès, B.; Bouillet, B. HDL Functionality in Type 1 Diabetes: Enhancement of Cholesterol Efflux Capacity in Relationship with Decreased HDL Carbamylation after Improvement of Glycemic Control. Cardiovasc. Diabetol. 2022, 21, 154. [Google Scholar] [CrossRef]
- Hadfield, K.A.; Pattison, D.I.; Brown, B.E.; Hou, L.; Rye, K.-A.; Davies, M.J.; Hawkins, C.L. Myeloperoxidase-Derived Oxidants Modify Apolipoprotein A-I and Generate Dysfunctional High-Density Lipoproteins: Comparison of Hypothiocyanous Acid (HOSCN) with Hypochlorous Acid (HOCl). Biochem. J. 2013, 449, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Zangger, K.; El-Gamal, D.; Binder, V.; Curcic, S.; Konya, V.; Schuligoi, R.; Heinemann, A.; Marsche, G. Myeloperoxidase-Derived Chlorinating Species Induce Protein Carbamylation through Decomposition of Thiocyanate and Urea: Novel Pathways Generating Dysfunctional High-Density Lipoprotein. Antioxid. Redox Signal. 2012, 17, 1043–1052. [Google Scholar] [CrossRef]
- Kheniser, K.G.; Osme, A.; Kim, C.; Ilchenko, S.; Kasumov, T.; Kashyap, S.R. Temporal Dynamics of High-Density Lipoprotein Proteome in Diet-Controlled Subjects with Type 2 Diabetes. Biomolecules 2020, 10, 520. [Google Scholar] [CrossRef]
- Duvillard, L.; Pont, F.; Florentin, E.; Gambert, P.; Vergès, B. Inefficiency of Insulin Therapy to Correct Apolipoprotein A-I Metabolic Abnormalities in Non-Insulin-Dependent Diabetes Mellitus. Atherosclerosis 2000, 152, 229–237. [Google Scholar] [CrossRef]
- Frénais, R.; Ouguerram, K.; Maugeais, C.; Mahot, P.; Maugère, P.; Krempf, M.; Magot, T. High Density Lipoprotein Apolipoprotein AI Kinetics in NIDDM: A Stable Isotope Study. Diabetologia 1997, 40, 578–583. [Google Scholar] [CrossRef]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef]
- Pont, F.; Duvillard, L.; Florentin, E.; Gambert, P.; Vergès, B. High-Density Lipoprotein Apolipoprotein A-I Kinetics in Obese Insulin Resistant Patients. An in Vivo Stable Isotope Study. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1151–1158. [Google Scholar] [CrossRef]
- Chan, D.C.; Barrett, P.H.R.; Ooi, E.M.M.; Ji, J.; Chan, D.T.; Watts, G.F. Very Low Density Lipoprotein Metabolism and Plasma Adiponectin as Predictors of High-Density Lipoprotein Apolipoprotein A-I Kinetics in Obese and Nonobese Men. J. Clin. Endocrinol. Metab. 2009, 94, 989–997. [Google Scholar] [CrossRef]
- Vergès, B.; Adiels, M.; Boren, J.; Barrett, P.H.; Watts, G.F.; Chan, D.; Duvillard, L.; Söderlund, S.; Matikainen, N.; Kahri, J.; et al. Interrelationships between the Kinetics of VLDL Subspecies and HDL Catabolism in Abdominal Obesity: A Multicenter Tracer Kinetic Study. J. Clin. Endocrinol. Metab. 2014, 99, 4281–4290. [Google Scholar] [CrossRef]
- Sparks, D.L.; Davidson, W.S.; Lund-Katz, S.; Phillips, M.C. Effects of the Neutral Lipid Content of High Density Lipoprotein on Apolipoprotein A-I Structure and Particle Stability. J. Biol. Chem. 1995, 270, 26910–26917. [Google Scholar] [CrossRef]
- Horowitz, B.S.; Goldberg, I.J.; Merab, J.; Vanni, T.M.; Ramakrishnan, R.; Ginsberg, H.N. Increased Plasma and Renal Clearance of an Exchangeable Pool of Apolipoprotein A-I in Subjects with Low Levels of High Density Lipoprotein Cholesterol. J. Clin. Investig. 1993, 91, 1743–1752. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, B.; Uffelman, K.D.; Carpentier, A.; Cohn, J.S.; Steiner, G.; Barrett, P.H.; Lewis, G.F. Triglyceride Enrichment of HDL Enhances in Vivo Metabolic Clearance of HDL Apo A-I in Healthy Men. J. Clin. Investig. 1999, 103, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B.; Adiels, M.; Boren, J.; Barrett, P.H.; Watts, G.F.; Chan, D.; Duvillard, L.; Söderlund, S.; Matikainen, N.; Kahri, J.; et al. ApoA-II HDL Catabolism and Its Relationships With the Kinetics of ApoA-I HDL and of VLDL1, in Abdominal Obesity. J. Clin. Endocrinol. Metab. 2016, 101, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Garçon, D.; Berger, J.-M.; Cariou, B.; Le May, C. Transintestinal Cholesterol Excretion in Health and Disease. Curr. Atheroscler. Rep. 2022, 24, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Ebtehaj, S.; Gruppen, E.G.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. HDL (High-Density Lipoprotein) Cholesterol Efflux Capacity Is Associated with Incident Cardiovascular Disease in the General Population. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1874–1883. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL Cholesterol Efflux Capacity with Incident Coronary Heart Disease Events: A Prospective Case-Control Study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Guerin, M.; Silvain, J.; Gall, J.; Darabi, M.; Berthet, M.; Frisdal, E.; Hauguel-Moreau, M.; Zeitouni, M.; Kerneis, M.; Lattuca, B.; et al. Association of Serum Cholesterol Efflux Capacity With Mortality in Patients With ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 3259–3269. [Google Scholar] [CrossRef]
- De Vries, R.; Groen, A.K.; Perton, F.G.; Dallinga-Thie, G.M.; van Wijland, M.J.A.; Dikkeschei, L.D.; Wolffenbuttel, B.H.R.; van Tol, A.; Dullaart, R.P.F. Increased Cholesterol Efflux from Cultured Fibroblasts to Plasma from Hypertriglyceridemic Type 2 Diabetic Patients: Roles of Pre β-HDL, Phospholipid Transfer Protein and Cholesterol Esterification. Atherosclerosis 2008, 196, 733–741. [Google Scholar] [CrossRef]
- Yassine, H.N.; Belopolskaya, A.; Schall, C.; Stump, C.S.; Lau, S.S.; Reaven, P.D. Enhanced Cholesterol Efflux to HDL through the ABCA1 Transporter in Hypertriglyceridemia of Type 2 Diabetes. Metabolism 2014, 63, 727–734. [Google Scholar] [CrossRef]
- Cavallero, E.; Brites, F.; Delfly, B.; Nicolaïew, N.; Decossin, C.; De Geitere, C.; Fruchart, J.C.; Wikinski, R.; Jacotot, B.; Castro, G. Abnormal Reverse Cholesterol Transport in Controlled Type II Diabetic Patients. Studies on Fasting and Postprandial LpA-I Particles. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2130–2135. [Google Scholar] [CrossRef]
- Syvänne, M.; Castro, G.; Dengremont, C.; De Geitere, C.; Jauhiainen, M.; Ehnholm, C.; Michelagnoli, S.; Franceschini, G.; Kahri, J.; Taskinen, M.R. Cholesterol Efflux from Fu5AH Hepatoma Cells Induced by Plasma of Subjects with or without Coronary Artery Disease and Non-Insulin-Dependent Diabetes: Importance of LpA-I:A-II Particles and Phospholipid Transfer Protein. Atherosclerosis 1996, 127, 245–253. [Google Scholar] [CrossRef]
- Zhou, H.; Shiu, S.W.; Wong, Y.; Tan, K.C. Impaired Serum Capacity to Induce Cholesterol Efflux Is Associated with Endothelial Dysfunction in Type 2 Diabetes Mellitus. Diabetes Vasc. Dis. Res. 2009, 6, 238–243. [Google Scholar] [CrossRef]
- Passarelli, M.; Shimabukuro, A.F.; Catanozi, S.; Nakandakare, E.R.; Rocha, J.C.; Carrilho, A.J.; Quintão, E.C. Diminished Rate of Mouse Peritoneal Macrophage Cholesterol Efflux Is Not Related to the Degree of HDL Glycation in Diabetes Mellitus. Clin. Chim. Acta 2000, 301, 119–134. [Google Scholar] [CrossRef]
- Murakami, H.; Tanabe, J.; Tamasawa, N.; Matsumura, K.; Yamashita, M.; Matsuki, K.; Murakami, H.; Matsui, J.; Suda, T. Reduction of Paraoxonase-1 Activity May Contribute the Qualitative Impairment of HDL Particles in Patients with Type 2 Diabetes. Diabetes Res. Clin. Pract. 2013, 99, 30–38. [Google Scholar] [CrossRef]
- He, Y.; Ronsein, G.E.; Tang, C.; Jarvik, G.P.; Davidson, W.S.; Kothari, V.; Song, H.D.; Segrest, J.P.; Bornfeldt, K.E.; Heinecke, J.W. Diabetes Impairs Cellular Cholesterol Efflux From ABCA1 to Small HDL Particles. Circ. Res. 2020, 127, 1198–1210. [Google Scholar] [CrossRef]
- Barberio, M.D.; Kasselman, L.J.; Playford, M.P.; Epstein, S.B.; Renna, H.A.; Goldberg, M.; DeLeon, J.; Voloshyna, I.; Barlev, A.; Salama, M.; et al. Cholesterol Efflux Alterations in Adolescent Obesity: Role of Adipose-Derived Extracellular Vesical MicroRNAs. J. Transl. Med. 2019, 17, 232. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Thaxton, C.S.; Berry, J.; Daviglus, M.L.; Yuan, C.; Sun, J.; Ayers, C.; Lloyd-Jones, D.M.; Wilkins, J.T. HDL Efflux Capacity, HDL Particle Size, and High-Risk Carotid Atherosclerosis in a Cohort of Asymptomatic Older Adults: The Chicago Healthy Aging Study. J. Lipid Res. 2017, 58, 600–606. [Google Scholar] [CrossRef]
- Alenezi, M.Y.; Marcil, M.; Blank, D.; Sherman, M.; Genest, J. Is the Decreased High-Density Lipoprotein Cholesterol in the Metabolic Syndrome Due to Cellular Lipid Efflux Defect? J. Clin. Endocrinol. Metab. 2004, 89, 761–764. [Google Scholar] [CrossRef]
- Annema, W.; Dikkers, A.; de Boer, J.F.; van Greevenbroek, M.M.J.; van der Kallen, C.J.H.; Schalkwijk, C.G.; Stehouwer, C.D.A.; Dullaart, R.P.F.; Tietge, U.J.F. Impaired HDL Cholesterol Efflux in Metabolic Syndrome Is Unrelated to Glucose Tolerance Status: The CODAM Study. Sci. Rep. 2016, 6, 27367. [Google Scholar] [CrossRef]
- Lucero, D.; Sviridov, D.; Svidirov, D.; Freeman, L.; López, G.I.; Fassio, E.; Remaley, A.T.; Schreier, L. Increased Cholesterol Efflux Capacity in Metabolic Syndrome: Relation with Qualitative Alterations in HDL and LCAT. Atherosclerosis 2015, 242, 236–242. [Google Scholar] [CrossRef]
- Gomes Kjerulf, D.; Wang, S.; Omer, M.; Pathak, A.; Subramanian, S.; Han, C.Y.; Tang, C.; den Hartigh, L.J.; Shao, B.; Chait, A. Glycation of HDL Blunts Its Anti-Inflammatory and Cholesterol Efflux Capacities in Vitro, but Has No Effect in Poorly Controlled Type 1 Diabetes Subjects. J. Diabetes Complicat. 2020, 34, 107693. [Google Scholar] [CrossRef] [PubMed]
- Rashduni, D.L.; Rifici, V.A.; Schneider, S.H.; Khachadurian, A.K. Glycation of High-Density Lipoprotein Does Not Increase Its Susceptibility to Oxidation or Diminish Its Cholesterol Efflux Capacity. Metabolism 1999, 48, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Duell, P.B.; Oram, J.F.; Bierman, E.L. Nonenzymatic Glycosylation of HDL and Impaired HDL-Receptor-Mediated Cholesterol Efflux. Diabetes 1991, 40, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, K.; Tamasawa, N.; Yamashita, M.; Tanabe, J.; Murakami, H.; Matsui, J.; Imaizumi, T.; Satoh, K.; Suda, T. Metformin Restores Impaired HDL-Mediated Cholesterol Efflux Due to Glycation. Atherosclerosis 2009, 206, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Espín, J.; Nilsson, O.; Bernfur, K.; Del Giudice, R.; Lagerstedt, J.O. Site-Specific Glycations of Apolipoprotein A-I Lead to Differentiated Functional Effects on Lipid-Binding and on Glucose Metabolism. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 2822–2834. [Google Scholar] [CrossRef]
- Shao, B.; Pennathur, S.; Pagani, I.; Oda, M.N.; Witztum, J.L.; Oram, J.F.; Heinecke, J.W. Modifying Apolipoprotein A-I by Malondialdehyde, but Not by an Array of Other Reactive Carbonyls, Blocks Cholesterol Efflux by the ABCA1 Pathway. J. Biol. Chem. 2010, 285, 18473–18484. [Google Scholar] [CrossRef]
- Huang, J.; Tao, H.; Yancey, P.G.; Leuthner, Z.; May-Zhang, L.S.; Jung, J.-Y.; Zhang, Y.; Ding, L.; Amarnath, V.; Liu, D.; et al. Scavenging Dicarbonyls with 5′-O-Pentyl-Pyridoxamine Increases HDL Net Cholesterol Efflux Capacity and Attenuates Atherosclerosis and Insulin Resistance. Mol. Metab. 2022, 67, 101651. [Google Scholar] [CrossRef]
- Shao, B.; Cavigiolio, G.; Brot, N.; Oda, M.N.; Heinecke, J.W. Methionine Oxidation Impairs Reverse Cholesterol Transport by Apolipoprotein A-I. Proc. Natl. Acad. Sci. USA 2008, 105, 12224–12229. [Google Scholar] [CrossRef]
- Nobecourt, E.; Davies, M.J.; Brown, B.E.; Curtiss, L.K.; Bonnet, D.J.; Charlton, F.; Januszewski, A.S.; Jenkins, A.J.; Barter, P.J.; Rye, K.-A. The Impact of Glycation on Apolipoprotein A-I Structure and Its Ability to Activate Lecithin: Cholesterol Acyltransferase. Diabetologia 2007, 50, 643–653. [Google Scholar] [CrossRef]
- De la Llera Moya, M.; McGillicuddy, F.C.; Hinkle, C.C.; Byrne, M.; Joshi, M.R.; Nguyen, V.; Tabita-Martinez, J.; Wolfe, M.L.; Badellino, K.; Pruscino, L.; et al. Inflammation Modulates Human HDL Composition and Function in Vivo. Atherosclerosis 2012, 222, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Vaisar, T.; Tang, C.; Babenko, I.; Hutchins, P.; Wimberger, J.; Suffredini, A.F.; Heinecke, J.W. Inflammatory Remodeling of the HDL Proteome Impairs Cholesterol Efflux Capacity. J. Lipid Res. 2015, 56, 1519–1530. [Google Scholar] [CrossRef]
- Agarwala, A.P.; Rodrigues, A.; Risman, M.; McCoy, M.; Trindade, K.; Qu, L.; Cuchel, M.; Billheimer, J.; Rader, D.J. High-Density Lipoprotein (HDL) Phospholipid Content and Cholesterol Efflux Capacity Are Reduced in Patients With Very High HDL Cholesterol and Coronary Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1515–1519. [Google Scholar] [CrossRef]
- Yancey, P.G.; Kawashiri, M.; Moore, R.; Glick, J.M.; Williams, D.L.; Connelly, M.A.; Rader, D.J.; Rothblat, G.H. In Vivo Modulation of HDL Phospholipid Has Opposing Effects on SR-BI- and ABCA1-Mediated Cholesterol Efflux. J. Lipid Res. 2004, 45, 337–346. [Google Scholar] [CrossRef]
- Subbaiah, P.V.; Gesquiere, L.R.; Wang, K. Regulation of the Selective Uptake of Cholesteryl Esters from High Density Lipoproteins by Sphingomyelin. J. Lipid Res. 2005, 46, 2699–2705. [Google Scholar] [CrossRef]
- Tölle, M.; Pawlak, A.; Schuchardt, M.; Kawamura, A.; Tietge, U.J.; Lorkowski, S.; Keul, P.; Assmann, G.; Chun, J.; Levkau, B.; et al. HDL-Associated Lysosphingolipids Inhibit NAD(P)H Oxidase-Dependent Monocyte Chemoattractant Protein-1 Production. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1542–1548. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Annema, W.; Tio, R.A.; Tietge, U.J.F. The HDL Anti-Inflammatory Function Is Impaired in Myocardial Infarction and May Predict New Cardiac Events Independent of HDL Cholesterol. Clin. Chim. Acta 2014, 433, 34–38. [Google Scholar] [CrossRef]
- Jia, C.; Anderson, J.L.C.; Gruppen, E.G.; Lei, Y.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. High-Density Lipoprotein Anti-Inflammatory Capacity and Incident Cardiovascular Events. Circulation 2021, 143, 1935–1945. [Google Scholar] [CrossRef]
- Morgantini, C.; Natali, A.; Boldrini, B.; Imaizumi, S.; Navab, M.; Fogelman, A.M.; Ferrannini, E.; Reddy, S.T. Anti-Inflammatory and Antioxidant Properties of HDLs Are Impaired in Type 2 Diabetes. Diabetes 2011, 60, 2617–2623. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Wagner, A.C.; Jung, C.-L.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Watson, A.D.; Hama, S.; Navab, M.; Anantharamaiah, G.M.; et al. Anti-Inflammatory ApoA-I-Mimetic Peptides Bind Oxidized Lipids with Much Higher Affinity than Human ApoA-I. J. Lipid Res. 2008, 49, 2302–2311. [Google Scholar] [CrossRef]
- Ebtehaj, S.; Gruppen, E.G.; Parvizi, M.; Tietge, U.J.F.; Dullaart, R.P.F. The Anti-Inflammatory Function of HDL Is Impaired in Type 2 Diabetes: Role of Hyperglycemia, Paraoxonase-1 and Low Grade Inflammation. Cardiovasc. Diabetol. 2017, 16, 132. [Google Scholar] [CrossRef] [Green Version]
- Van Tienhoven-Wind, L.J.N.; Tietge, U.J.F.; Dullaart, R.P.F. The HDL Anti-Inflammatory Function Is Impaired in the Context of Low-Normal Free Thyroxine in Diabetic and Nondiabetic Individuals. Clin. Endocrinol. 2018, 88, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Frej, C.; Holmér, A.; Guo, L.J.; Tran, S.; Dahlbäck, B. High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Drew, B.G.; Nakhla, S.; Duffy, S.J.; Murphy, A.J.; Barter, P.J.; Rye, K.-A.; Chin-Dusting, J.; Hoang, A.; Sviridov, D.; et al. Reconstituted High-Density Lipoprotein Increases Plasma High-Density Lipoprotein Anti-Inflammatory Properties and Cholesterol Efflux Capacity in Patients with Type 2 Diabetes. J. Am. Coll. Cardiol. 2009, 53, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ji, L.; Zhang, D.; Tong, X.; Pan, B.; Liu, P.; Zhang, Y.; Huang, Y.; Su, J.; Willard, B.; et al. Nonenzymatic Glycation of High-Density Lipoprotein Impairs Its Anti-Inflammatory Effects in Innate Immunity. Diabetes Metab. Res. Rev. 2012, 28, 186–195. [Google Scholar] [CrossRef]
- Schwendeman, A.; Sviridov, D.O.; Yuan, W.; Guo, Y.; Morin, E.E.; Yuan, Y.; Stonik, J.; Freeman, L.; Ossoli, A.; Thacker, S.; et al. The Effect of Phospholipid Composition of Reconstituted HDL on Its Cholesterol Efflux and Anti-Inflammatory Properties. J. Lipid Res. 2015, 56, 1727–1737. [Google Scholar] [CrossRef]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms Underlying Adverse Effects of HDL on ENOS-Activating Pathways in Patients with Coronary Artery Disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef]
- Marshall, H.E.; Stamler, J.S. Inhibition of NF-Kappa B by S-Nitrosylation. Biochemistry 2001, 40, 1688–1693. [Google Scholar] [CrossRef]
- McGrath, K.C.Y.; Li, X.H.; Puranik, R.; Liong, E.C.; Tan, J.T.M.; Dy, V.M.; DiBartolo, B.A.; Barter, P.J.; Rye, K.A.; Heather, A.K. Role of 3beta-Hydroxysteroid-Delta 24 Reductase in Mediating Antiinflammatory Effects of High-Density Lipoproteins in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 877–882. [Google Scholar] [CrossRef]
- Wu, B.J.; Chen, K.; Shrestha, S.; Ong, K.L.; Barter, P.J.; Rye, K.-A. High-Density Lipoproteins Inhibit Vascular Endothelial Inflammation by Increasing 3β-Hydroxysteroid-Δ24 Reductase Expression and Inducing Heme Oxygenase-1. Circ. Res. 2013, 112, 278–288. [Google Scholar] [CrossRef]
- Cheng, A.M.; Handa, P.; Tateya, S.; Schwartz, J.; Tang, C.; Mitra, P.; Oram, J.F.; Chait, A.; Kim, F. Apolipoprotein A-I Attenuates Palmitate-Mediated NF-ΚB Activation by Reducing Toll-like Receptor-4 Recruitment into Lipid Rafts. PLoS ONE 2012, 7, e33917. [Google Scholar] [CrossRef] [Green Version]
- Dueñas, A.I.; Aceves, M.; Fernández-Pisonero, I.; Gómez, C.; Orduña, A.; Crespo, M.S.; García-Rodríguez, C. Selective Attenuation of Toll-like Receptor 2 Signalling May Explain the Atheroprotective Effect of Sphingosine 1-Phosphate. Cardiovasc. Res. 2008, 79, 537–544. [Google Scholar] [CrossRef]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative Activity of High-Density Lipoprotein (HDL): Mechanistic Insights into Potential Clinical Benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Elsøe, S.; Ahnström, J.; Christoffersen, C.; Hoofnagle, A.N.; Plomgaard, P.; Heinecke, J.W.; Binder, C.J.; Björkbacka, H.; Dahlbäck, B.; Nielsen, L.B. Apolipoprotein M Binds Oxidized Phospholipids and Increases the Antioxidant Effect of HDL. Atherosclerosis 2012, 221, 91–97. [Google Scholar] [CrossRef]
- Mastorikou, M.; Mackness, B.; Liu, Y.; Mackness, M. Glycation of Paraoxonase-1 Inhibits Its Activity and Impairs the Ability of High-Density Lipoprotein to Metabolize Membrane Lipid Hydroperoxides. Diabet. Med. 2008, 25, 1049–1055. [Google Scholar] [CrossRef]
- Nobécourt, E.; Jacqueminet, S.; Hansel, B.; Chantepie, S.; Grimaldi, A.; Chapman, M.J.; Kontush, A. Defective Antioxidative Activity of Small Dense HDL3 Particles in Type 2 Diabetes: Relationship to Elevated Oxidative Stress and Hyperglycaemia. Diabetologia 2005, 48, 529–538. [Google Scholar] [CrossRef]
- Hansel, B.; Giral, P.; Nobecourt, E.; Chantepie, S.; Bruckert, E.; Chapman, M.J.; Kontush, A. Metabolic Syndrome Is Associated with Elevated Oxidative Stress and Dysfunctional Dense High-Density Lipoprotein Particles Displaying Impaired Antioxidative Activity. J. Clin. Endocrinol. Metab. 2004, 89, 4963–4971. [Google Scholar] [CrossRef]
- Sanguinetti, S.M.; Brites, F.D.; Fasulo, V.; Verona, J.; Elbert, A.; Wikinski, R.L.; Schreier, L.E. HDL Oxidability and Its Protective Effect against LDL Oxidation in Type 2 Diabetic Patients. Diabetes Nutr. Metab. 2001, 14, 27–36. [Google Scholar]
- Brindisi, M.C.; Duvillard, L.; Monier, S.; Vergès, B.; Perségol, L. Deleterious Effect of Glycation on the Ability of HDL to Counteract the Inhibitory Effect of Oxidized LDL on Endothelium-Dependent Vasorelaxation. Diabetes Metab. Res. Rev. 2013, 29, 618–623. [Google Scholar] [CrossRef]
- Kontush, A.; de Faria, E.C.; Chantepie, S.; Chapman, M.J. A Normotriglyceridemic, Low HDL-Cholesterol Phenotype Is Characterised by Elevated Oxidative Stress and HDL Particles with Attenuated Antioxidative Activity. Atherosclerosis 2005, 182, 277–285. [Google Scholar] [CrossRef]
- Iwakiri, Y. S-Nitrosylation of Proteins: A New Insight into Endothelial Cell Function Regulated by ENOS-Derived NO. Nitric. Oxide 2011, 25, 95–101. [Google Scholar] [CrossRef]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-Density Lipoprotein Binding to Scavenger Receptor-BI Activates Endothelial Nitric Oxide Synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Westerterp, M.; Koetsveld, J.; Fernández-Hernando, C.; Yvan-Charvet, L.; Wang, N.; Sessa, W.C.; Tall, A.R. ATP-Binding Cassette Transporter G1 and High-Density Lipoprotein Promote Endothelial NO Synthesis through a Decrease in the Interaction of Caveolin-1 and Endothelial NO Synthase. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.-R.; van der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; et al. HDL Induces NO-Dependent Vasorelaxation via the Lysophospholipid Receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Yuhanna, I.S.; Quon, M.J.; Shaul, P.W. High Density Lipoprotein-Induced Endothelial Nitric-Oxide Synthase Activation Is Mediated by Akt and MAP Kinases. J. Biol. Chem. 2003, 278, 9142–9149. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.; Gräler, M.; Keul, P.; Weske, S.; Reimann, C.-M.; Jindrová, H.; Kleinbongard, P.; Sabbadini, R.; Bröcker-Preuss, M.; Erbel, R.; et al. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content. J. Am. Coll. Cardiol. 2015, 66, 1470–1485. [Google Scholar] [CrossRef]
- Ossoli, A.; Simonelli, S.; Varrenti, M.; Morici, N.; Oliva, F.; Stucchi, M.; Gomaraschi, M.; Strazzella, A.; Arnaboldi, L.; Thomas, M.J.; et al. Recombinant LCAT (Lecithin:Cholesterol Acyltransferase) Rescues Defective HDL (High-Density Lipoprotein)-Mediated Endothelial Protection in Acute Coronary Syndrome. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 915–924. [Google Scholar] [CrossRef]
- Ruiz, M.; Okada, H.; Dahlbäck, B. HDL-Associated ApoM Is Anti-Apoptotic by Delivering Sphingosine 1-Phosphate to S1P1 & S1P3 Receptors on Vascular Endothelium. Lipids Health Dis. 2017, 16, 36. [Google Scholar] [CrossRef]
- Nofer, J.R.; Levkau, B.; Wolinska, I.; Junker, R.; Fobker, M.; von Eckardstein, A.; Seedorf, U.; Assmann, G. Suppression of Endothelial Cell Apoptosis by High Density Lipoproteins (HDL) and HDL-Associated Lysosphingolipids. J. Biol. Chem. 2001, 276, 34480–34485. [Google Scholar] [CrossRef]
- De Souza, J.A.; Vindis, C.; Hansel, B.; Nègre-Salvayre, A.; Therond, P.; Serrano, C.V.; Chantepie, S.; Salvayre, R.; Bruckert, E.; Chapman, M.J.; et al. Metabolic Syndrome Features Small, Apolipoprotein A-I-Poor, Triglyceride-Rich HDL3 Particles with Defective Anti-Apoptotic Activity. Atherosclerosis 2008, 197, 84–94. [Google Scholar] [CrossRef]
- Sang, H.; Yao, S.; Zhang, L.; Li, X.; Yang, N.; Zhao, J.; Zhao, L.; Si, Y.; Zhang, Y.; Lv, X.; et al. Walk-Run Training Improves the Anti-Inflammation Properties of High-Density Lipoprotein in Patients with Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2015, 100, 870–879. [Google Scholar] [CrossRef]
- Kontush, A.; Therond, P.; Zerrad, A.; Couturier, M.; Négre-Salvayre, A.; de Souza, J.A.; Chantepie, S.; Chapman, M.J. Preferential Sphingosine-1-Phosphate Enrichment and Sphingomyelin Depletion Are Key Features of Small Dense HDL3 Particles: Relevance to Antiapoptotic and Antioxidative Activities. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1843–1849. [Google Scholar] [CrossRef] [Green Version]
- Noor, R.; Shuaib, U.; Wang, C.X.; Todd, K.; Ghani, U.; Schwindt, B.; Shuaib, A. High-Density Lipoprotein Cholesterol Regulates Endothelial Progenitor Cells by Increasing ENOS and Preventing Apoptosis. Atherosclerosis 2007, 192, 92–99. [Google Scholar] [CrossRef]
- Seo, M.H.; Bae, J.C.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; Park, S.W.; Kim, S.W.; Lee, W.-Y. Association of Lipid and Lipoprotein Profiles with Future Development of Type 2 Diabetes in Nondiabetic Korean Subjects: A 4-Year Retrospective, Longitudinal Study. J. Clin. Endocrinol. Metab. 2011, 96, E2050–E2054. [Google Scholar] [CrossRef]
- Abbasi, A.; Corpeleijn, E.; Gansevoort, R.T.; Gans, R.O.B.; Hillege, H.L.; Stolk, R.P.; Navis, G.; Bakker, S.J.L.; Dullaart, R.P.F. Role of HDL Cholesterol and Estimates of HDL Particle Composition in Future Development of Type 2 Diabetes in the General Population: The PREVEND Study. J. Clin. Endocrinol. Metab. 2013, 98, E1352–E1359. [Google Scholar] [CrossRef]
- Wilson, P.W.F.; Meigs, J.B.; Sullivan, L.; Fox, C.S.; Nathan, D.M.; D’Agostino, R.B. Prediction of Incident Diabetes Mellitus in Middle-Aged Adults: The Framingham Offspring Study. Arch. Intern. Med. 2007, 167, 1068–1074. [Google Scholar] [CrossRef]
- Hu, P.L.; Koh, Y.L.E.; Tan, N.C. The Utility of Diabetes Risk Score Items as Predictors of Incident Type 2 Diabetes in Asian Populations: An Evidence-Based Review. Diabetes Res. Clin. Pract. 2016, 122, 179–189. [Google Scholar] [CrossRef]
- White, J.; Swerdlow, D.I.; Preiss, D.; Fairhurst-Hunter, Z.; Keating, B.J.; Asselbergs, F.W.; Sattar, N.; Humphries, S.E.; Hingorani, A.D.; Holmes, M.V. Association of Lipid Fractions with Risks for Coronary Artery Disease and Diabetes. JAMA Cardiol. 2016, 1, 692–699. [Google Scholar] [CrossRef]
- Fall, T.; Xie, W.; Poon, W.; Yaghootkar, H.; Mägi, R.; Knowles, J.W.; Lyssenko, V.; Weedon, M.; Frayling, T.M.; the GENESIS Consortium; et al. Using Genetic Variants to Assess the Relationship Between Circulating Lipids and Type 2 Diabetes. Diabetes 2015, 64, 2676–2684. [Google Scholar] [CrossRef]
- Dangas, K.; Navar, A.-M.; Kastelein, J.J.P. The Effect of CETP Inhibitors on New-Onset Diabetes: A Systematic Review and Meta-Analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 622–632. [Google Scholar] [CrossRef]
- Cochran, B.J.; Ong, K.-L.; Manandhar, B.; Rye, K.-A. High Density Lipoproteins and Diabetes. Cells 2021, 10, 850. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Lindahl, M.; Petrlova, J.; Dalla-Riva, J.; Göransson, O.; Cushman, S.W.; Krupinska, E.; Jones, H.A.; Lagerstedt, J.O. Single Injections of ApoA-I Acutely Improve in Vivo Glucose Tolerance in Insulin-Resistant Mice. Diabetologia 2014, 57, 797–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-Density Lipoprotein Modulates Glucose Metabolism in Patients with Type 2 Diabetes Mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.; Del Giudice, R.; Nagao, M.; Grönberg, C.; Eliasson, L.; Lagerstedt, J.O. Apolipoprotein A-I Primes Beta Cells to Increase Glucose Stimulated Insulin Secretion. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165613. [Google Scholar] [CrossRef] [PubMed]
- Pétremand, J.; Puyal, J.; Chatton, J.-Y.; Duprez, J.; Allagnat, F.; Frias, M.; James, R.W.; Waeber, G.; Jonas, J.-C.; Widmann, C. HDLs Protect Pancreatic β-Cells against ER Stress by Restoring Protein Folding and Trafficking. Diabetes 2012, 61, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tabet, F.; Cochran, B.J.; Cuesta Torres, L.F.; Wu, B.J.; Barter, P.J.; Rye, K.-A. Apolipoprotein A-I Enhances Insulin-Dependent and Insulin-Independent Glucose Uptake by Skeletal Muscle. Sci. Rep. 2019, 9, 1350. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Duparc, T.; Ruidavets, J.-B.; Genoux, A.; Ingueneau, C.; Najib, S.; Ferrières, J.; Perret, B.; Martinez, L.O. Serum Level of HDL Particles Are Independently Associated with Long-Term Prognosis in Patients with Coronary Artery Disease: The GENES Study. Sci. Rep. 2020, 10, 8138. [Google Scholar] [CrossRef]
- Murakami, K.; Harada, A.; Toh, R.; Kubo, T.; Miwa, K.; Kim, J.; Kiriyama, M.; Iino, T.; Nishikawa, Y.; Uno, S.-N.; et al. Fully Automated Immunoassay for Cholesterol Uptake Capacity to Assess High-Density Lipoprotein Function and Cardiovascular Disease Risk. Sci. Rep. 2023, 13, 1899. [Google Scholar] [CrossRef]
- Hansel, B.; Bonnefont-Rousselot, D.; Orsoni, A.; Bittar, R.; Giral, P.; Roussel, R.; Marre, M.; Mohammedi, K.; Bruckert, E.; Chapman, M.J.; et al. Lifestyle Intervention Enhances High-Density Lipoprotein Function among Patients with Metabolic Syndrome Only at Normal Low-Density Lipoprotein Cholesterol Plasma Levels. J. Clin. Lipidol. 2016, 10, 1172–1181. [Google Scholar] [CrossRef]
- Takata, K.; Imaizumi, S.; Kawachi, E.; Suematsu, Y.; Shimizu, T.; Abe, S.; Matsuo, Y.; Tsukahara, H.; Noda, K.; Yahiro, E.; et al. Impact of Cigarette Smoking Cessation on High-Density Lipoprotein Functionality. Circ. J. 2014, 78, 2955–2962. [Google Scholar] [CrossRef]
- Kawachi, E.; Takata, K.; Imaizumi, S.; Miura, S.; Saku, K. Effects of Smoking Cessation on HDL Functionality. Tob. Induc. Dis. 2019, 17, A42. [Google Scholar] [CrossRef]
- Machado, A.P.; Pinto, R.S.; Moysés, Z.P.; Nakandakare, E.R.; Quintão, E.C.R.; Passarelli, M. Aminoguanidine and Metformin Prevent the Reduced Rate of HDL-Mediated Cell Cholesterol Efflux Induced by Formation of Advanced Glycation End Products. Int. J. Biochem. Cell Biol. 2006, 38, 392–403. [Google Scholar] [CrossRef]
- He, X.; Chen, X.; Wang, L.; Wang, W.; Liang, Q.; Yi, L.; Wang, Y.; Gao, Q. Metformin Ameliorates Ox-LDL-Induced Foam Cell Formation in Raw264.7 Cells by Promoting ABCG-1 Mediated Cholesterol Efflux. Life Sci. 2019, 216, 67–74. [Google Scholar] [CrossRef]
- Luo, F.; Guo, Y.; Ruan, G.-Y.; Long, J.-K.; Zheng, X.-L.; Xia, Q.; Zhao, S.-P.; Peng, D.-Q.; Fang, Z.-F.; Li, X.-P. Combined Use of Metformin and Atorvastatin Attenuates Atherosclerosis in Rabbits Fed a High-Cholesterol Diet. Sci. Rep. 2017, 7, 2169. [Google Scholar] [CrossRef]
- Perségol, L.; Duvillard, L.; Monier, S.; Brindisi, M.-C.; Bouillet, B.; Petit, J.-M.; Vergès, B. No Improvement of High-Density Lipoprotein (HDL) Vasorelaxant Effect despite Increase in HDL Cholesterol Concentration in Type 2 Diabetic Patients Treated with Glitazones. J. Clin. Endocrinol. Metab. 2014, 99, E2015–E2019. [Google Scholar] [CrossRef]
- Osto, E.; Doytcheva, P.; Corteville, C.; Bueter, M.; Dörig, C.; Stivala, S.; Buhmann, H.; Colin, S.; Rohrer, L.; Hasballa, R.; et al. Rapid and Body Weight-Independent Improvement of Endothelial and High-Density Lipoprotein Function after Roux-En-Y Gastric Bypass: Role of Glucagon-like Peptide-1. Circulation 2015, 131, 871–881. [Google Scholar] [CrossRef]
- Fadini, G.P.; Bonora, B.M.; Zatti, G.; Vitturi, N.; Iori, E.; Marescotti, M.C.; Albiero, M.; Avogaro, A. Effects of the SGLT2 Inhibitor Dapagliflozin on HDL Cholesterol, Particle Size, and Cholesterol Efflux Capacity in Patients with Type 2 Diabetes: A Randomized Placebo-Controlled Trial. Cardiovasc. Diabetol. 2017, 16, 42. [Google Scholar] [CrossRef]
- Ooi, E.M.M.; Watts, G.F.; Nestel, P.J.; Sviridov, D.; Hoang, A.; Barrett, P.H.R. Dose-Dependent Regulation of High-Density Lipoprotein Metabolism with Rosuvastatin in the Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 430–437. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Nguyen, M.N.; Barrett, P.H.R. Factorial Study of the Effect of N-3 Fatty Acid Supplementation and Atorvastatin on the Kinetics of HDL Apolipoproteins A-I and A-II in Men with Abdominal Obesity. Am. J. Clin. Nutr. 2006, 84, 37–43. [Google Scholar] [CrossRef]
- Triolo, M.; Annema, W.; de Boer, J.F.; Tietge, U.J.F.; Dullaart, R.P.F. Simvastatin and Bezafibrate Increase Cholesterol Efflux in Men with Type 2 Diabetes. Eur. J. Clin. Investig. 2014, 44, 240–248. [Google Scholar] [CrossRef]
- Maranghi, M.; Hiukka, A.; Badeau, R.; Sundvall, J.; Jauhiainen, M.; Taskinen, M.-R. Macrophage Cholesterol Efflux to Plasma and HDL in Subjects with Low and High Homocysteine Levels: A FIELD Substudy. Atherosclerosis 2011, 219, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, M.; Giammanco, A.; Purrello, F.; Pavanello, C.; Mombelli, G.; Di Pino, A.; Piro, S.; Cefalù, A.B.; Calabresi, L.; Averna, M.; et al. Effects of PCSK9 Inhibitors on HDL Cholesterol Efflux and Serum Cholesterol Loading Capacity in Familial Hypercholesterolemia Subjects: A Multi-Lipid-Center Real-World Evaluation. Front. Mol. Biosci. 2022, 9, 925587. [Google Scholar] [CrossRef] [PubMed]
- Metzinger, M.P.; Saldanha, S.; Gulati, J.; Patel, K.V.; El-Ghazali, A.; Deodhar, S.; Joshi, P.H.; Ayers, C.; Rohatgi, A. Effect of Anacetrapib on Cholesterol Efflux Capacity: A Substudy of the DEFINE Trial. J. Am. Hearth Assoc. 2020, 9, e018136. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ray, K.K.; Ballantyne, C.M.; Beacham, L.A.; Miller, D.L.; Ruotolo, G.; Nissen, S.E.; Riesmeyer, J.S.; ACCENTUATE Investigators. Comparative Effects of Cholesteryl Ester Transfer Protein Inhibition, Statin or Ezetimibe on Lipid Factors: The ACCENTUATE Trial. Atherosclerosis 2017, 261, 12–18. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Adam, S.; Ho, J.H.; Liu, Y.; Siahmansur, T.; Siddals, K.; Iqbal, Z.; Azmi, S.; Senapati, S.; New, J.; Jeziorska, M.; et al. Bariatric Surgery-Induced High-Density Lipoprotein Functionality Enhancement Is Associated with Reduced Inflammation. J. Clin. Endocrinol. Metab. 2022, 107, 2182–2194. [Google Scholar] [CrossRef]
- Thakkar, H.; Vincent, V.; Shukla, S.; Sra, M.; Kanga, U.; Aggarwal, S.; Singh, A. Improvements in Cholesterol Efflux Capacity of HDL and Adiponectin Contribute to Mitigation in Cardiovascular Disease Risk after Bariatric Surgery in a Cohort with Morbid Obesity. Diabetol. Metab. Syndr. 2021, 13, 46. [Google Scholar] [CrossRef]
- Lorkowski, S.W.; Brubaker, G.; Rotroff, D.M.; Kashyap, S.R.; Bhatt, D.L.; Nissen, S.E.; Schauer, P.R.; Aminian, A.; Smith, J.D. Bariatric Surgery Improves HDL Function Examined by ApoA1 Exchange Rate and Cholesterol Efflux Capacity in Patients with Obesity and Type 2 Diabetes. Biomolecules 2020, 10, 551. [Google Scholar] [CrossRef]
- Genua, I.; Puig, N.; Miñambres, I.; Benítez, S.; Gil, P.; Grau-Agramunt, M.; Rivas-Urbina, A.; Balagué, C.; Fernández-Alanin, S.; García-Osuna, Á.; et al. Changes in the Composition and Function of Lipoproteins after Bariatric Surgery in Patients with Severe Obesity. J. Clin. Med. 2021, 10, 1716. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Subjects 1 | Cell Model | Acceptor | CEC in Patients |
|---|---|---|---|---|
| Alenezi [120] | 22 MetS/9 controls Glucose: 5.7 ± 1.4 mmol/L TG: 3.84 ± 1.77 mmol/L | Human fibroblasts | Purified apoA-I | No difference in cholesterol and phospholipids efflux |
| Annema [121] | 297 MetS/255 non-MetS HbA1c: 6.2 ± 0.9% TG: 1.90 (1.40–2.20) mmol/L | Human THP-1 macrophages | ApoB-depleted serum | No difference for T2DM 7% decrease in MetS patients |
| Apro [18] | 35 T2DM/35 controls | Human THP-1 macrophages | HDLs from plasma and interstitial fluid | 10% decrease (plasma) 28% decrease (interstitial fluid) |
| Cavallero [112] | 14 T2DM/12 controls HbA1c: 5.1 ± 0.9% TG: 2.05 ± 0.73 mmol/L | Ob 1771 preadipocyte | LpA-I 2 | 50% decrease |
| Denimal [41] | 20 T2DM/25 controls HbA1c: 10.0 ± 2.3% TG: 2.38 ± 1.01 mmol/L | Human THP-1 macrophages | ApoB-depleted serum | No difference |
| Dullaart [28] | 76 MetS/94 controls Glucose: 8.4 ± 2.6 mmol/L TG: 1.35 mmol/L | Human fibroblasts | Whole plasma | 3.5% increase |
| Dullaart [30] | 75 T2DM/75 controls HbA1c: 6.7 ± 1.0% TG: 1.73 (1.17–2.17) mmol/L | Human fibroblasts | Whole plasma | No difference |
| Feng [81] | 6 T2DM/6 controls HbA1c: 10.9 ± 1.3% TG: 2.2 ± 0.8 mmol/L | Human THP-1 macrophages | HDLs | No difference |
| He [117] | 19 T2DM/20 controls HbA1c: 7.2 ± 1.2% TG: 1.47 ± 0.44 mmol/L | Baby hamster kidney (BHK) cells | Fractioned HDLs | 23% decrease in ABCA1-mediated CEC of small HDLs |
| Kashyap [97] | 9 T2DM/8 controls HbA1c: 6.3 ± 0.3% TG: 1.13 ± 0.42 mmol/L | Murine RAW264.7 macrophages | ApoB-depleted serum | Decrease in ABCA1-mediated and total CEC |
| Low [27] | 26 T2DM/26 controls HbA1c: 7.7 ± 1.2% TG: 1.7 ± 0.8 mmol/L | Human THP-1 macrophages | Whole plasma, apoB-depleted serum, HDLs | 30% increase (whole plasma), 34% increase (apoB-depleted serum), 50% increase (HDLs) |
| Lucero [122] | 35 MetS/15 controls Glucose: 7.4 ± 3.3 mmol/L TG: 1.73 ± 0.62 mmol/L | ABCA1 or ABCG1- transfected BHK cells | ApoB-depleted serum | 19% increase in ABCA1-mediated CEC |
| Murakami [116] | 36 T2DM/9 controls HbA1c: 9.5 ± 1.7% TG: 1.49 ± 0.77 mmol/L | Human THP-1 macrophages | HDLs | 7.3% decrease in CEC |
| Passarelli [115] | 18 T2DM/26 controls HbA1c: 12 ± 2% TG: 2.64 ± 1.51 mmol/L | Murine peritoneal macrophages | HDL subfractions | >50% decrease in HDL3-mediated CEC |
| Shiu [34] | 172 T2DM/175 controls HbA1c: 8.2 ± 1.4% TG: 1.40 (0.90–2.10) mmol/L | Fu5AH hepatoma cells | Whole serum | 9% decrease in SR-B1-mediated CEC. No difference in ABCA1-mediated CEC. |
| Shiu [36] | 180 T2DM/120 controls | Fu5AH hepatoma cells | Whole serum | 17% decrease in ABCA1-mediated CEC |
| Syvänne [113] | 100 T2DM/81 controls TG: 2.15 ± 1.20 mmol/L | Fu5AH hepatoma cells | Whole plasma | Reduction in CEC |
| Tsun [68] | 264 T2DM/275 controls HbA1c: 8.5 ± 1.7% TG: 1.60 (1.2–2.2) mmol/L | Fu5AH ABCG1-transfected CHO cells | Whole serum | Reduction in ABCG1- and SR-B1- mediated CEC |
| Yassine [111] | 45 T2DM with hypertriglyceridemia/ 26 T2DM without hypertriglyceridemia HbA1c: 8 ± 2%/8 ± 2% TG: 2.55 ± 0.99/1.03 ± 0.25 mmol/L | BHK | ApoB-depleted serum | 14% increase in ABCA1-dependent CEC in T2DM patients with hypertriglyceridemia |
| Zhou [37] | 60 T2DM/20 controls HbA1c: 8.3 ± 0.3% TG: 1.4 (1.0–2.6) mmol/L | Fu5AH hepatoma cells | Whole serum | 20 and 14% reduction in ABCA1-mediated and SR-B1-mediated CEC, respectively |
| Zhou [114] | 137 T2DM/75 controls HbA1c: 8.0 ± 1.3% TG: 1.50 (1.00–1.90) mmol/L | Fu5AH hepatoma cells | Whole serum | 7% reduction |
| Study | Subjects 1 | Cell Model | Results in Patients |
|---|---|---|---|
| Chen [87] | 102 T2DM with CAD/46 T2DM without CAD/40 controls HbA1c: 7.7 ± 1.3% TG: 1.77 ± 1.15 mmol/L | HUVECs and monocytes | Increased adhesion of monocytes to HUVECs in T2DM patients with CAD compared to controls. |
| Denimal [41] | 20 T2DM/25 controls HbA1c: 10.0 ± 2.3% TG: 2.38 ± 1.01 mmol/L | HUVECs | No difference in the ability of T2DM HDLs to inhibit VCAM-1 gene expression compared to control HDLs |
| Ebtehaj [142] | 40 T2DM/36 controls HbA1c: 6.7 ± 2.9% TG: 1.67 (1.22–2.16) mmol/L | HUVECs | Decreased ability of T2DM HDLs to inhibit VCAM-1 gene expression |
| Liu [146] | 6 T2DM/6 controls HbA1c: 10.8 ± 1.12% TG: 2.05 ± 0.21 mmol/L | Human THP-1 macrophages | Decreased ability of T2DM apoA-I to inhibit the LPS-induced release of TNF-α and IL-1β |
| Liu [73] | 6 T2DM/6 controls HbA1c: 10.8 ± 1.12% TG: 2.05 ± 0.21 mmol/L | Human THP-1 macrophages | Decreased ability of T2DM apoA-I to inhibit the release of TNF-α and IL-1β |
| Morgantini [140] | 93 T2DM/31 controls HbA1c: 8 ± 2% TG: 1.93 ± 1.54 mmol/L | HAECs and monocytes | Reduction in ability of T2DM HDLs to inhibit LDL-induced migration of monocytes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denimal, D.; Monier, S.; Bouillet, B.; Vergès, B.; Duvillard, L. High-Density Lipoprotein Alterations in Type 2 Diabetes and Obesity. Metabolites 2023, 13, 253. https://doi.org/10.3390/metabo13020253
Denimal D, Monier S, Bouillet B, Vergès B, Duvillard L. High-Density Lipoprotein Alterations in Type 2 Diabetes and Obesity. Metabolites. 2023; 13(2):253. https://doi.org/10.3390/metabo13020253
Chicago/Turabian StyleDenimal, Damien, Serge Monier, Benjamin Bouillet, Bruno Vergès, and Laurence Duvillard. 2023. "High-Density Lipoprotein Alterations in Type 2 Diabetes and Obesity" Metabolites 13, no. 2: 253. https://doi.org/10.3390/metabo13020253
APA StyleDenimal, D., Monier, S., Bouillet, B., Vergès, B., & Duvillard, L. (2023). High-Density Lipoprotein Alterations in Type 2 Diabetes and Obesity. Metabolites, 13(2), 253. https://doi.org/10.3390/metabo13020253