
Deregulated Metabolic Pathways in Ovarian Cancer: Cause and Consequence

 ,
,
Abstract
:1. Introduction
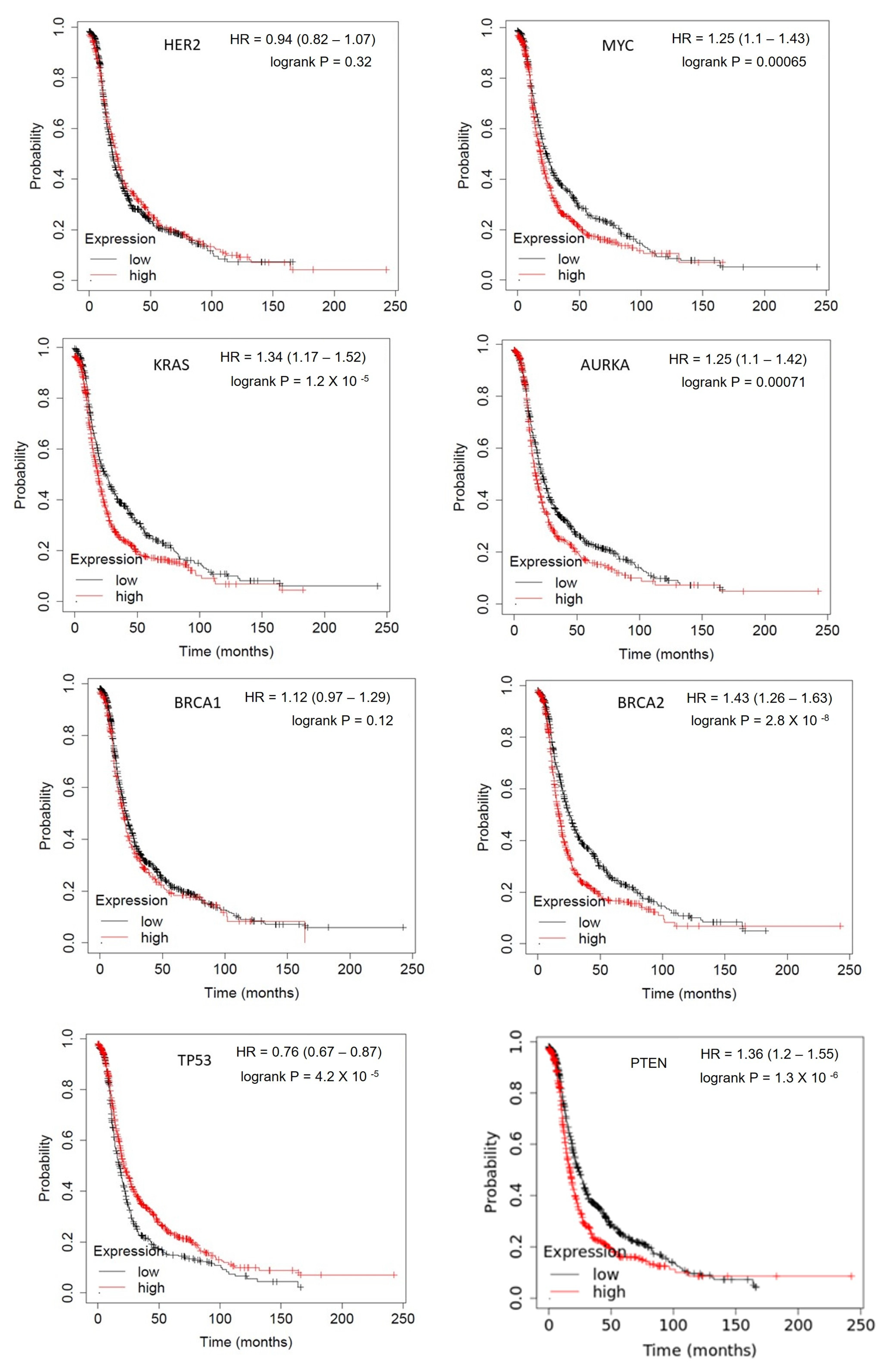
1.1. Somatic Driver Mutations
1.1.1. Her-2/neu
1.1.2. c-MYC
1.1.3. KRAS
1.1.4. BTAK
1.1.5. TP53
1.1.6. BRCA1
1.1.7. BRCA2
1.1.8. PTEN
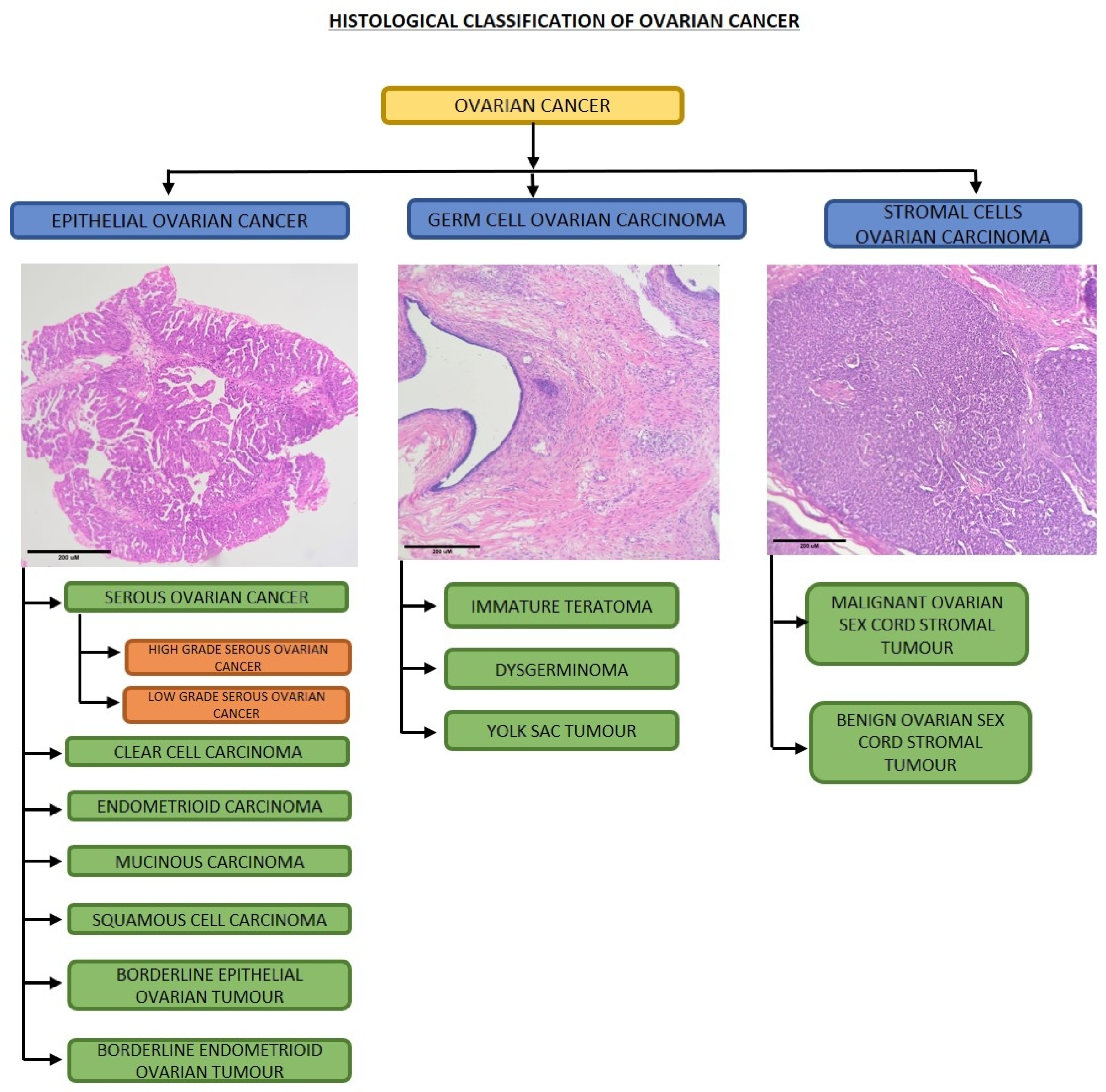
2. Deregulated Metabolic Pathways in Ovarian Cancer
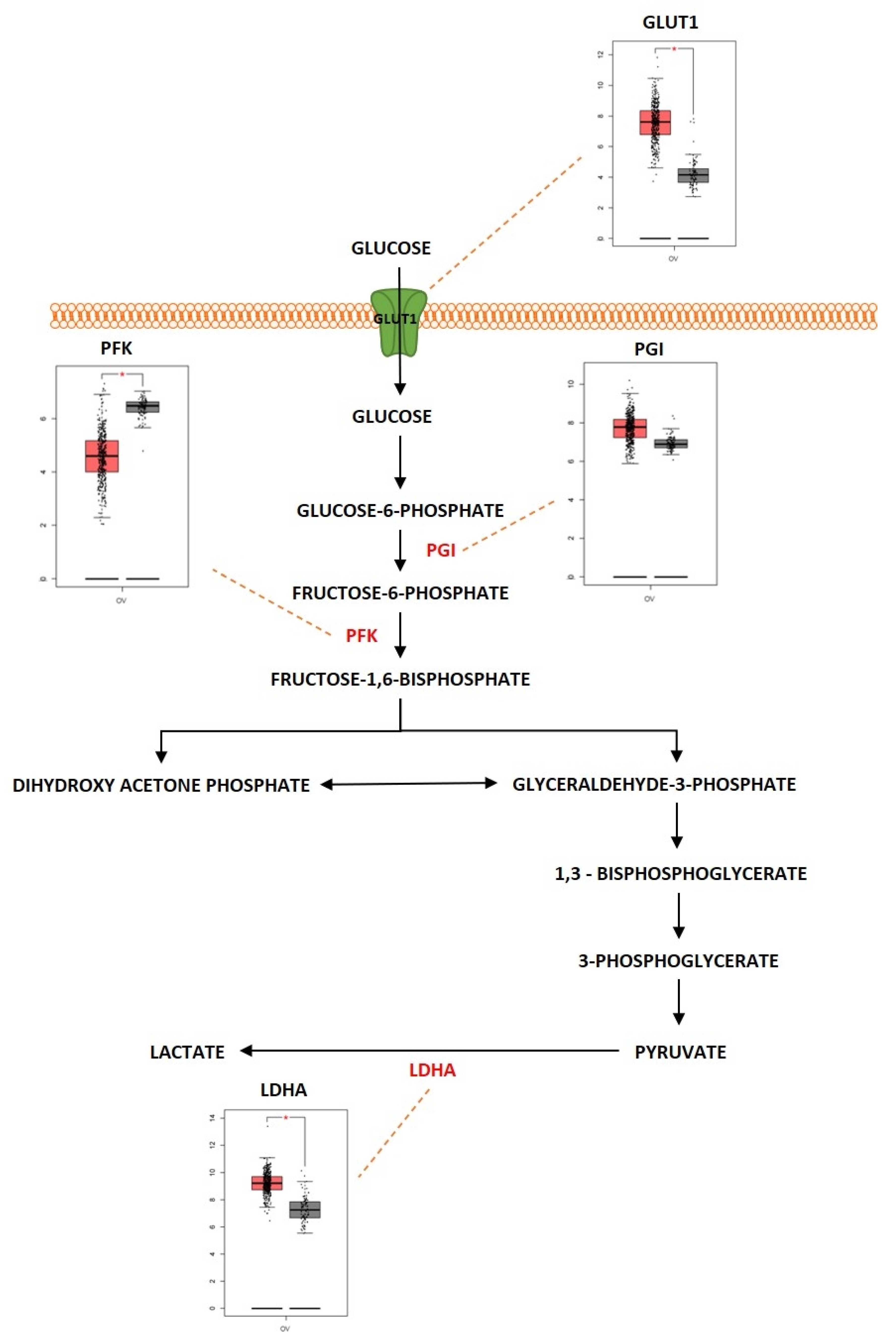
2.1. Glycolysis
Outcome of Somatic Driver Mutations in Glycolysis
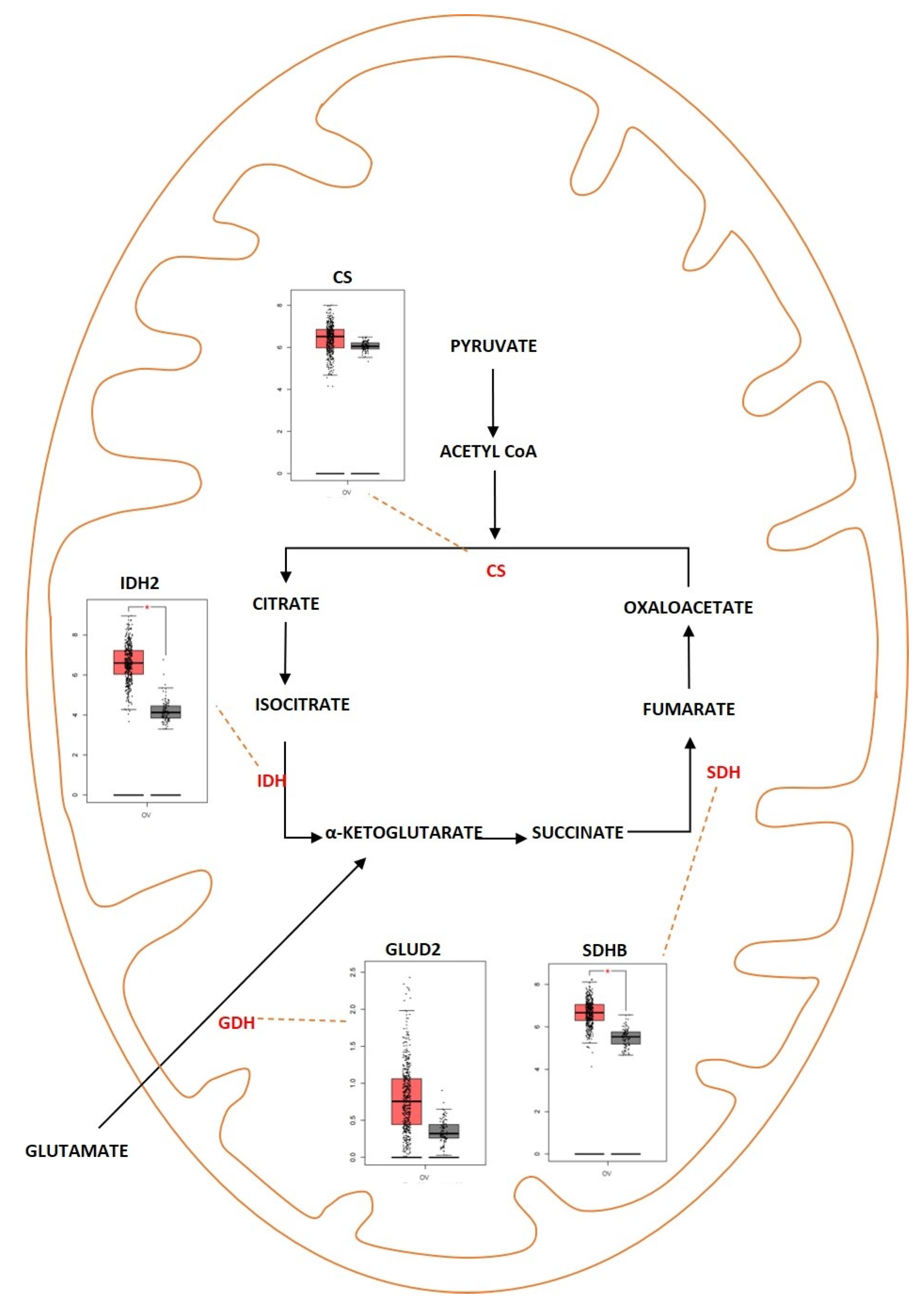
2.2. Tricarboxylic Acid Cycle
Outcome of Somatic Driver Mutations in the TCA Cycle
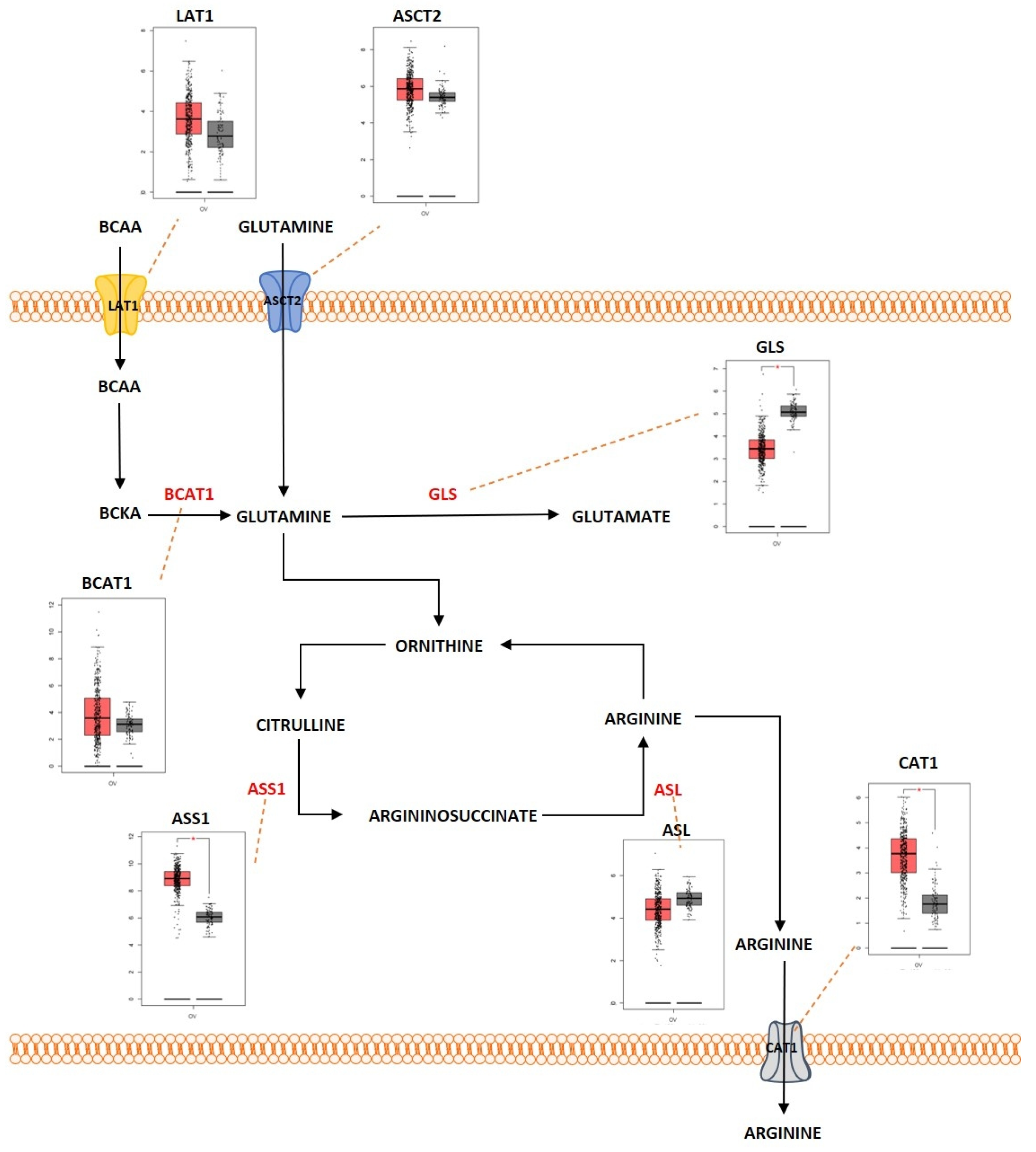
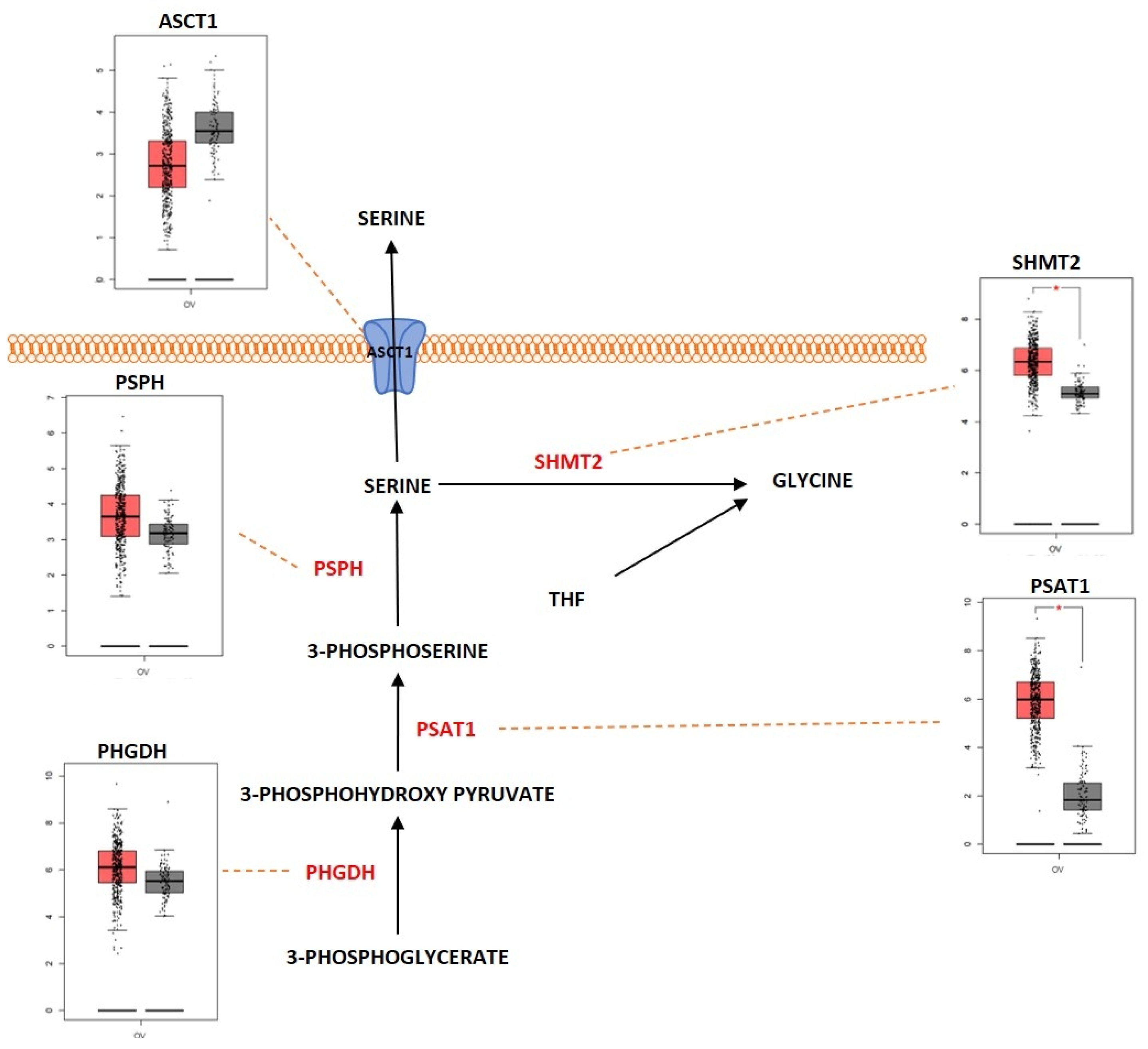
2.3. Amino Acid Metabolism
Outcome of Somatic Driver Mutations in Amino Acid Metabolism
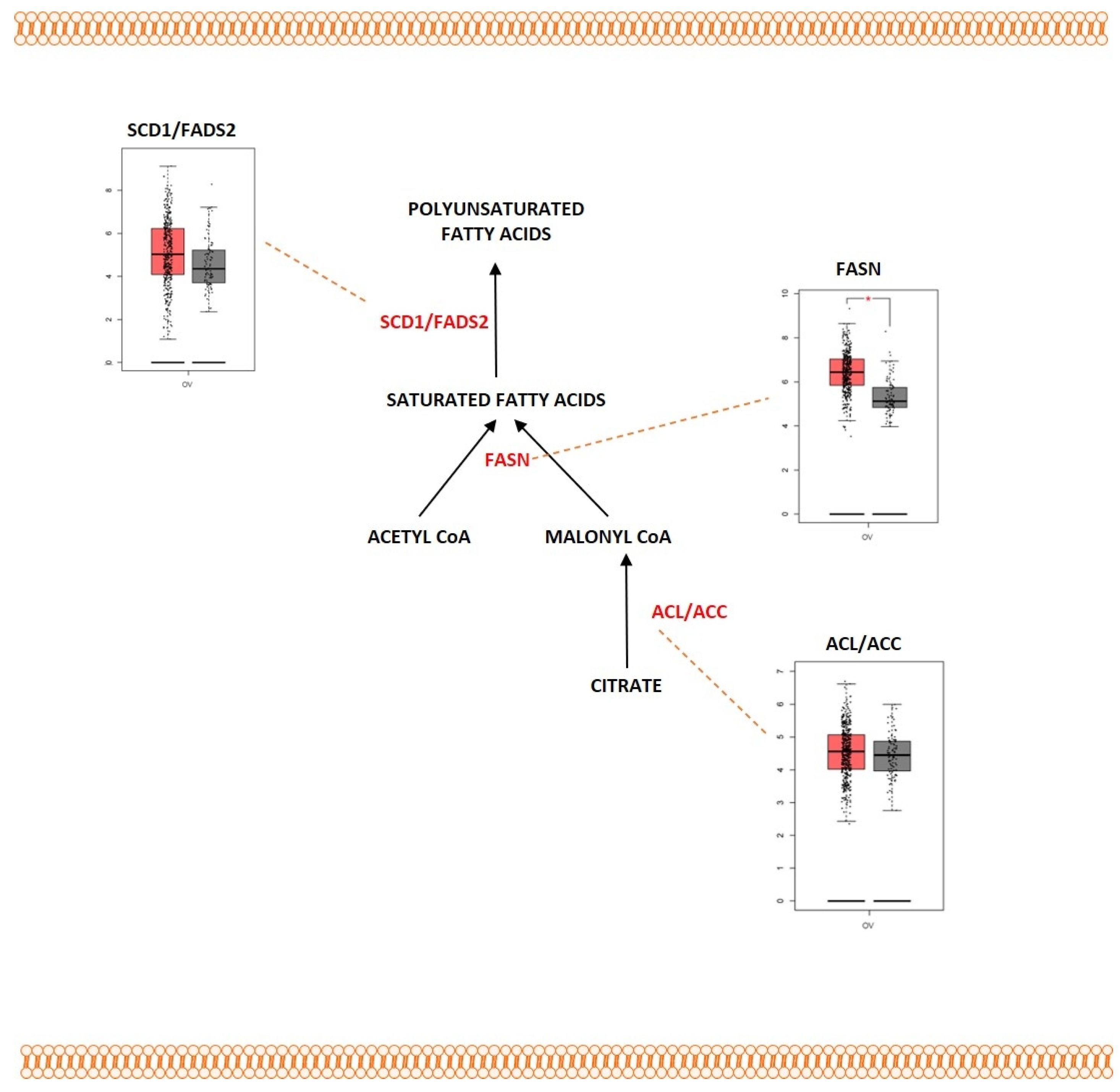
2.4. Fatty Acid Metabolism
Outcome of Somatic Driver Mutations in Fatty Acid Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Metabolic Proteins | Differential Expression | References |
|---|---|---|---|
| Glycolysis | GLUT1 | Overexpression | [73,74] |
| HK2 | Overexpression | [77] | |
| PFK | Overexpression | [89,90] | |
| LDHA | Overexpression | [80,81] | |
| Tricarboxylic acid cycle | IDH1 | Overexpression | [118] |
| SDHB | Reduced expression | [125,126] | |
| CS | Overexpression | [127] | |
| Amino acid metabolism | LAT1 | Overexpression | [168,193] |
| ASCT2 | Overexpression | [144,145] | |
| BCAT1 | Overexpression | [197,198] | |
| GLS | Overexpression | [147,148] | |
| ASS1 | Overexpression | [187] | |
| BCKDK | Overexpression | [199] | |
| CAT1 | Overexpression | [182] | |
| ASCT1 | Overexpression | [168] | |
| SHMT2 | Overexpression | [178] | |
| PSAT1 | Overexpression | [175] | |
| PHGDH | Overexpression | [173] | |
| Fatty acid metabolism | CD36 | Overexpression | |
| FABP4 | Overexpression | [221] | |
| ACLY | Overexpression | [236,237,238] | |
| ACC | Overexpression | [236,237,238] | |
| FASN | Overexpression | [226,227,228] | |
| SCD1 | Overexpression | [229] | |
| FADS2 | Overexpression | [231] |
3. FDA-Approved Drugs Targeting Ovarian Cancer and Their Role in Cancer Metabolism
3.1. Chemotherapeutic Drugs
3.2. Monoclonal Antibodies
3.3. Small Molecule Inhibitors
| Class of the Drug | FDA-Approved Drug | Role of the Drug in Cancer Metabolism | References |
|---|---|---|---|
| Chemotherapeutic drugs | Melphalan | Melphalan efficiently targeted the DNA repair mechanisms in ovarian cancer patients with BRCA1/2 mutations. | [247,248,249] |
| Carboplatin | Carboplatin in combination with PFK158 promoted lipophagy in chemoresistant cells. | [251] | |
| Cisplatin | Cisplatin interacted with GSH and regulated ROS homeostasis. Cisplatin redirected the cancer cells from the aerobic glycolysis to oxidative phosphorylation. Cisplatin downregulated HK2 and PDK. | [253,254,255,256,257,258] | |
| Cyclophosphamide | Cyclophosphamide was identified to trigger ferroptosis by increasing the ROS and intracellular iron levels and by decreasing GSH levels. | [261] | |
| Doxorubicin | Doxorubicin stimulated fatty acid oxidation and inhibit de novo lipogenesis by the activation of p53. Doxorubicin induced ROS in the mitochondria, with the help of NADPH oxidase enzyme. | [263,264] | |
| Gemcitabine hydrochloride | The overexpression of the FASN enzyme promoted resistance in cancer cells that were treated with gemcitabine. | [266] | |
| Paclitaxel | Paclitaxel decreased the levels of glucose-1,6-bisphosphate and fructose-1,6-bisphosphate and caused the detachment of phosphofructokinase from the cytoskeleton of cancer cells. | [269] | |
| Topotecan hydrochloride | Topotecan in combination with DT-13 inhibited HK2 activity which in turn suppressed aerobic glycolysis. | [271] | |
| Monoclonal antibodies | Bevacizumab | Bevacizumab induced lipid metabolic rewiring and upregulated pathways that are involved in lipid metabolism in ovarian cancer. | [273] |
| Mirvetuximab soravtansine-gynx | Mirvetuximab deregulated folate metabolism by binding and blocking FOLR1 receptor. | [275] | |
| Small molecule inhibitors | Olaparib | Enacted metabolic reprogramming of glutamine-derived amino acids and lipid metabolism in Olaparib-treated cancer cells. Olaparib caused metabolic shift from anaerobic glycolysis to tricarboxylic acid cycle which was induced resulting in increased ATP synthesis. | [278,281,282] |
| Niraparib tosylate | Niraparib-adapted tumor cells had lesser levels of stored glucose as GSK3 phosphorylates the enzyme glycogen synthase. Niraparib induced a metabolic shift to tri-carboxylic acid cycle from anaerobic glycolysis which resulted in increased ATP synthesis. | [280,281,282] |
4. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Khazaei, Z.; Namayandeh, S.M.; Beiranvand, R.; Naemi, H.; Bechashk, S.M.; Goodarzi, E. Worldwide incidence and mortality of ovarian cancer and Human Development Index (HDI): GLOBOCAN sources and methods 2018. J. Prev. Med. Hyg. 2021, 62, E174–E184. [Google Scholar] [PubMed]
- Gajjar, K.; Ogden, G.; Mujahid, M.I.; Razvi, K. Symptoms and risk factors of ovarian cancer: A survey in primary care. ISRN Obstet. Gynecol. 2012, 2012, 754197. [Google Scholar] [CrossRef] [Green Version]
- Mok, S.C.; Kwong, J.; Welch, W.R.; Samimi, G.; Ozbun, L.; Bonome, T.; Birrer, M.J.; Berkowitz, R.S.; Wong, K.K. Etiology and pathogenesis of epithelial ovarian cancer. Dis. Markers 2007, 23, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortolero-Luna, G.; Mitchell, M.F. The epidemiology of ovarian cancer. J. Cell Biochem. Suppl. 1995, 23, 200–207. [Google Scholar] [CrossRef]
- Kisielewski, R.; Tolwinska, A.; Mazurek, A.; Laudanski, P. Inflammation and ovarian cancer—Current views. Ginekol. Pol. 2013, 84, 293–297. [Google Scholar] [CrossRef]
- Wei, J.J.; William, J.; Bulun, S. Endometriosis and ovarian cancer: A review of clinical, pathologic, and molecular aspects. Int. J. Gynecol. Pathol. 2011, 30, 553–568. [Google Scholar] [CrossRef] [Green Version]
- Eoh, K.J.; Kim, J.E.; Park, H.S.; Lee, S.T.; Park, J.S.; Han, J.W.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, J.H.; et al. Detection of Germline Mutations in Patients with Epithelial Ovarian Cancer Using Multi-gene Panels: Beyond BRCA1/2. Cancer Res. Treat. 2018, 50, 917–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukomska, A.; Menkiszak, J.; Gronwald, J.; Tomiczek-Szwiec, J.; Szwiec, M.; Jasiowka, M.; Blecharz, P.; Kluz, T.; Stawicka-Nielacna, M.; Madry, R.; et al. Recurrent Mutations in BRCA1, BRCA2, RAD51C, PALB2 and CHEK2 in Polish Patients with Ovarian Cancer. Cancers 2021, 13, 849. [Google Scholar] [CrossRef]
- Nakamura, K.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Nomura, H.; Hirasawa, A.; et al. Features of ovarian cancer in Lynch syndrome (Review). Mol. Clin. Oncol. 2014, 2, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Landen, C.N., Jr.; Birrer, M.J.; Sood, A.K. Early events in the pathogenesis of epithelial ovarian cancer. J. Clin. Oncol. 2008, 26, 995–1005. [Google Scholar] [CrossRef]
- Shih Ie, M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Saegusa, M.; Hashimura, M.; Yoshida, T.; Okayasu, I. beta- Catenin mutations and aberrant nuclear expression during endometrial tumorigenesis. Br. J. Cancer 2001, 84, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.O.; et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Nakayama, N.; Kurman, R.J.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.E.; Wang, T.L.; Shih, I.M. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol. Ther. 2006, 5, 779–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Lim, H.J.; Ledger, W. Targeted therapy in ovarian cancer. Womens Health 2016, 12, 363–378. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B. Discovery and ranking of the most robust prognostic biomarkers in serous ovarian cancer. Geroscience 2023. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Koopman, T.; van der Vegt, B.; Dijkstra, M.; Bart, J.; Duiker, E.; Wisman, G.B.A.; de Bock, G.H.; Hollema, H. HER2 immunohistochemistry in endometrial and ovarian clear cell carcinoma: Discordance between antibodies and with in-situ hybridisation. Histopathology 2018, 73, 852–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankaj, S.; Kumari, J.; Choudhary, V.; Kumari, A.; Kumari, S.; Kumari, A.; Nazneen, S.; Madhawi, R.; Kumar, S. Prognostic Value of HER-2/neu Gene Amplification in Epithelial Ovarian Carcinoma. J. Obstet. Gynaecol. India 2019, 69, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, H.; Ye, Q.; Wang, C.; Xu, Y. Prognostic Value of KIF2A and HER2-Neu Overexpression in Patients With Epithelial Ovarian Cancer. Medicine 2016, 95, e2803. [Google Scholar] [CrossRef] [PubMed]
- Dimova, I.; Raitcheva, S.; Dimitrov, R.; Doganov, N.; Toncheva, D. Correlations between c-myc gene copy-number and clinicopathological parameters of ovarian tumours. Eur. J. Cancer 2006, 42, 674–679. [Google Scholar] [CrossRef]
- Yamamoto, A.; Kurata, M.; Yamamoto, K.; Nogawa, D.; Inoue, M.; Ishibashi, S.; Ikeda, M.; Miyasaka, N.; Kitagawa, M. High amplification of PVT1 and MYC predict favorable prognosis in early ovarian carcinoma. Pathol. Res. Pract. 2020, 216, 153175. [Google Scholar] [CrossRef]
- Yasue, H.; Takeda, A.; Ishibashi, M. Amplification of the c-myc gene and the elevation of its transcripts in human ovarian tumor lines. Cell Struct. Funct. 1987, 12, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Auner, V.; Kriegshauser, G.; Tong, D.; Horvat, R.; Reinthaller, A.; Mustea, A.; Zeillinger, R. KRAS mutation analysis in ovarian samples using a high sensitivity biochip assay. BMC Cancer 2009, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Cuatrecasas, M.; Villanueva, A.; Matias-Guiu, X.; Prat, J. K-ras mutations in mucinous ovarian tumors: A clinicopathologic and molecular study of 95 cases. Cancer 1997, 79, 1581–1586. [Google Scholar] [CrossRef]
- Mandai, M.; Konishi, I.; Kuroda, H.; Komatsu, T.; Yamamoto, S.; Nanbu, K.; Matsushita, K.; Fukumoto, M.; Yamabe, H.; Mori, T. Heterogeneous distribution of K-ras-mutated epithelia in mucinous ovarian tumors with special reference to histopathology. Hum. Pathol. 1998, 29, 34–40. [Google Scholar] [CrossRef]
- Gritsko, T.M.; Coppola, D.; Paciga, J.E.; Yang, L.; Sun, M.; Shelley, S.A.; Fiorica, J.V.; Nicosia, S.V.; Cheng, J.Q. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin. Cancer Res. 2003, 9, 1420–1426. [Google Scholar] [PubMed]
- Landen, C.N., Jr.; Lin, Y.G.; Immaneni, A.; Deavers, M.T.; Merritt, W.M.; Spannuth, W.A.; Bodurka, D.C.; Gershenson, D.M.; Brinkley, W.R.; Sood, A.K. Overexpression of the centrosomal protein Aurora—A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin. Cancer Res. 2007, 13, 4098–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, M.Q.; Baba, T.; Lee, P.S.; Barnett, J.C.; Sfakianos, G.P.; Secord, A.A.; Murphy, S.K.; Iversen, E.; Marks, J.R.; Berchuck, A. Expression signatures of TP53 mutations in serous ovarian cancers. BMC Cancer 2010, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Saretzki, G.; Hoffmann, U.; Rohlke, P.; Psille, R.; Gaigal, T.; Keller, G.; Hofler, H.; Loning, T.; Petersen, I.; Dietel, M. Identification of allelic losses in benign, borderline, and invasive epithelial ovarian tumors and correlation with clinical outcome. Cancer 1997, 80, 1241–1249. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study, G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Stratton, J.F.; Gayther, S.A.; Russell, P.; Dearden, J.; Gore, M.; Blake, P.; Easton, D.; Ponder, B.A. Contribution of BRCA1 mutations to ovarian cancer. N. Engl. J. Med. 1997, 336, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; Tone, A.A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod. Pathol. 2014, 27, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Obata, K.; Morland, S.J.; Watson, R.H.; Hitchcock, A.; Chenevix-Trench, G.; Thomas, E.J.; Campbell, I.G. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998, 58, 2095–2097. [Google Scholar]
- Shih, C.; Padhy, L.C.; Murray, M.; Weinberg, R.A. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 1981, 290, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M.M. HER2 Amplification in Tumors Activates PI3K/Akt Signaling Independent of HER3. Cancer Res. 2018, 78, 3645–3658. [Google Scholar] [CrossRef] [Green Version]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.J.; Barrett, J.; Villa-Garcia, M.; Dang, C.V. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 1990, 10, 5914–5920. [Google Scholar] [CrossRef]
- Popescu, N.C.; Amsbaugh, S.C.; DiPaolo, J.A.; Tronick, S.R.; Aaronson, S.A.; Swan, D.C. Chromosomal localization of three human ras genes by in situ molecular hybridization. Somat. Cell Mol. Genet. 1985, 11, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Rubinfeld, B.; Albert, I.; McCormick, F. RapV12 antagonizes Ras-dependent activation of ERK1 and ERK2 by LPA and EGF in Rat-1 fibroblasts. EMBO J. 1993, 12, 3475–3485. [Google Scholar] [CrossRef] [PubMed]
- de Vries-Smits, A.M.; Burgering, B.M.; Leevers, S.J.; Marshall, C.J.; Bos, J.L. Involvement of p21ras in activation of extracellular signal-regulated kinase 2. Nature 1992, 357, 602–604. [Google Scholar] [CrossRef]
- Sen, S.; Zhou, H.; White, R.A. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene 1997, 14, 2195–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Chang, B.; Yang, F.; Guo, X.; Cai, K.Q.; Xiao, X.S.; Wang, H.; Sen, S.; Hung, M.C.; Mills, G.B.; et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin. Cancer Res. 2010, 16, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, O.W.; Merry, D.; Givol, D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13). Proc. Natl. Acad. Sci. USA 1986, 83, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plisiecka-Halasa, J.; Dansonka-Mieszkowska, A.; Kraszewska, E.; Danska-Bidzinska, A.; Kupryjanczyk, J. Loss of heterozygosity, microsatellite instability and TP53 gene status in ovarian carcinomas. Anticancer Res. 2008, 28, 989–996. [Google Scholar]
- Zhang, G.N.; Liu, H.; Huang, J.M.; Wang, L.; Zhao, J.S.; Li, C.; Mi, K.; Zhu, Y.; Cheng, J.; Zha, X. TP53 K351N mutation-associated platinum resistance after neoadjuvant chemotherapy in patients with advanced ovarian cancer. Gynecol. Oncol. 2014, 132, 752–757. [Google Scholar] [CrossRef]
- Tuna, M.; Ju, Z.; Yoshihara, K.; Amos, C.I.; Tanyi, J.L.; Mills, G.B. Clinical relevance of TP53 hotspot mutations in high-grade serous ovarian cancers. Br. J. Cancer 2020, 122, 405–412. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Simard, J.; Rommens, J.; Couch, F.; Shattuck-Eidens, D.; Neuhausen, S.; Merajver, S.; Thorlacius, S.; Offit, K.; Stoppa-Lyonnet, D.; et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat. Genet. 1996, 12, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Frattini, M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front. Oncol. 2013, 3, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Teng, Y.; Zhang, Y.; Qu, K.; Yang, X.; Fu, J.; Chen, W.; Li, X. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget 2015, 6, 40799–40814. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Mandai, M.; Matsumura, N.; Yamaguchi, K.; Kondoh, H.; Amano, Y.; Baba, T.; Hamanishi, J.; Abiko, K.; Kosaka, K.; et al. Hepatocyte nuclear factor-1beta (HNF-1beta) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol. Carcinog. 2015, 54, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Caneba, C.A.; Yang, L.; Baddour, J.; Curtis, R.; Win, J.; Hartig, S.; Marini, J.; Nagrath, D. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014, 5, e1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.Y.; Temkin, S.M.; Fang, X. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yun, Y.; Wu, B.; Wen, L.; Wen, M.; Yang, H.; Zhao, L.; Liu, W.; Huang, S.; Wen, N.; et al. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget 2016, 7, 47985–47997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.Y.; Yang, Y.; Xie, J.Y.; Lu, Y.L.; Shi, K.; Huang, Y.Q. MicroRNA-144 mediates metabolic shift in ovarian cancer cells by directly targeting Glut1. Tumour Biol. 2016, 37, 6855–6860. [Google Scholar] [CrossRef]
- Siu, M.K.Y.; Jiang, Y.X.; Wang, J.J.; Leung, T.H.Y.; Han, C.Y.; Tsang, B.K.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. Hexokinase 2 Regulates Ovarian Cancer Cell Migration, Invasion and Stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 Signaling Cascades. Cancers 2019, 11, 813. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wang, L.; Chen, W.; Wang, Y.; Zhen, S.; Chen, H.; Cheng, J.; Zhou, Y.; Li, X.; Zhao, L. miR-603 targeted hexokinase-2 to inhibit the malignancy of ovarian cancer cells. Arch. Biochem. Biophys. 2019, 661, 1–9. [Google Scholar] [CrossRef]
- Lu, J.; Zhen, S.; Tuo, X.; Chang, S.; Yang, X.; Zhou, Y.; Chen, W.; Zhao, L.; Li, X. Downregulation of DNMT3A Attenuates the Warburg Effect, Proliferation, and Invasion via Promoting the Inhibition of miR-603 on HK2 in Ovarian Cancer. Technol. Cancer Res. Treat. 2022, 21, 15330338221110668. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Kontomanolis, E.; Giatromanolaki, A.; Sivridis, E.; Liberis, V. Serum and tissue LDH levels in patients with breast/gynaecological cancer and benign diseases. Gynecol. Obstet. Investig. 2009, 67, 162–168. [Google Scholar] [CrossRef]
- Qiu, H.; Jackson, A.L.; Kilgore, J.E.; Zhong, Y.; Chan, L.L.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget 2015, 6, 6915–6930. [Google Scholar] [CrossRef] [Green Version]
- Donadeu, F.X.; Schauer, S.N. Differential miRNA expression between equine ovulatory and anovulatory follicles. Domest. Anim. Endocrinol. 2013, 45, 122–125. [Google Scholar] [CrossRef]
- Han, R.L.; Wang, F.P.; Zhang, P.A.; Zhou, X.Y.; Li, Y. miR-383 inhibits ovarian cancer cell proliferation, invasion and aerobic glycolysis by targeting LDHA. Neoplasma 2017, 64, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. [Google Scholar] [CrossRef] [Green Version]
- Holloway, R.W.; Marignani, P.A. Targeting mTOR and Glycolysis in HER2-Positive Breast Cancer. Cancers 2021, 13, 2922. [Google Scholar] [CrossRef]
- Choi, J.; Jung, W.H.; Koo, J.S. Metabolism-related proteins are differentially expressed according to the molecular subtype of invasive breast cancer defined by surrogate immunohistochemistry. Pathobiology 2013, 80, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, E.S.; Koo, J.S. Expression of Pentose Phosphate Pathway-Related Proteins in Breast Cancer. Dis. Markers 2018, 2018, 9369358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Deng, Y.; Zhu, J.; Duan, Y.; Weng, W.; Wu, X. Pim1 promotes cell proliferation and regulates glycolysis via interaction with MYC in ovarian cancer. Onco Targets Ther. 2018, 11, 6647–6656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racker, E.; Resnick, R.J.; Feldman, R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc. Natl. Acad. Sci. USA 1985, 82, 3535–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Shitara, M.; Yokota, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. Overexpression of GLUT1 correlates with Kras mutations in lung carcinomas. Mol. Med. Rep. 2012, 5, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Qin, Y.; Zhang, B.; Ji, S.; Shi, S.; Xu, W.; Liu, J.; Xiang, J.; Liang, D.; Hu, Q.; et al. ARF6, induced by mutant Kras, promotes proliferation and Warburg effect in pancreatic cancer. Cancer Lett. 2017, 388, 303–311. [Google Scholar] [CrossRef]
- Connell, L.C.; Boucher, T.M.; Chou, J.F.; Capanu, M.; Maldonado, S.; Kemeny, N.E. Relevance of CEA and LDH in relation to KRAS status in patients with unresectable colorectal liver metastases. J. Surg. Oncol. 2017, 115, 480–487. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics 2020, 10, 6928–6945. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Shang, E.; Shu, C.; Kim, S.; Mela, A.; Humala, N.; Mahajan, A.; Yang, H.W.; Akman, H.O.; Quinzii, C.M.; et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat. Commun. 2021, 12, 5203. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Shang, E.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells 2022, 11, 2956. [Google Scholar] [CrossRef]
- Cheng, A.; Zhang, P.; Wang, B.; Yang, D.; Duan, X.; Jiang, Y.; Xu, T.; Jiang, Y.; Shi, J.; Ding, C.; et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat. Commun. 2019, 10, 5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, T.; Kusuyama, J.; Bandow, K.; Matsuguchi, T. Glut1 expression is increased by p53 reduction to switch metabolism to glycolysis during osteoblast differentiation. Biochem. J. 2020, 477, 1795–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, E.; Pedersen, P.L. High aerobic glycolysis of rat hepatoma cells in culture: Role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. USA 1977, 74, 3735–3739. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Chiyoda, T.; Hart, P.C.; Eckert, M.A.; McGregor, S.M.; Lastra, R.R.; Hamamoto, R.; Nakamura, Y.; Yamada, S.D.; Olopade, O.I.; Lengyel, E.; et al. Loss of BRCA1 in the Cells of Origin of Ovarian Cancer Induces Glycolysis: A Window of Opportunity for Ovarian Cancer Chemoprevention. Cancer Prev. Res. 2017, 10, 255–266. [Google Scholar] [CrossRef] [Green Version]
- van der Groep, P.; Bouter, A.; Menko, F.H.; van der Wall, E.; van Diest, P.J. High frequency of HIF-1alpha overexpression in BRCA1 related breast cancer. Breast Cancer Res. Treat. 2008, 111, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.J.; Morvan, D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE 2014, 9, e102438. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.C.; McBrayer, S.K.; Coarfa, C.; Dalva-Aydemir, S.; Gunaratne, P.H.; Carpten, J.D.; Keats, J.K.; Rosen, S.T.; Shanmugam, M. Expression and phosphorylation of the AS160_v2 splice variant supports GLUT4 activation and the Warburg effect in multiple myeloma. Cancer Metab. 2013, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morani, F.; Phadngam, S.; Follo, C.; Titone, R.; Aimaretti, G.; Galetto, A.; Alabiso, O.; Isidoro, C. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J. Mol. Endocrinol. 2014, 53, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7, 84999–85020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta 2012, 1826, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Wang, J.; Zhang, L.; Wu, D.; Yu, D.; Tian, X.; Liu, J.; Jiang, X.; Shen, Y.; Zhang, L.; et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 2015, 32, 391. [Google Scholar] [CrossRef] [Green Version]
- Bergaggio, E.; Piva, R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 2019, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Sonego, M.; Baldassarre, G. A new role for IDH1 in the control of ovarian cancer cells metabolism and senescence. Ann. Transl. Med. 2020, 8, 780. [Google Scholar] [CrossRef]
- Dahl, E.S.; Buj, R.; Leon, K.E.; Newell, J.M.; Imamura, Y.; Bitler, B.G.; Snyder, N.W.; Aird, K.M. Targeting IDH1 as a Prosenescent Therapy in High-grade Serous Ovarian Cancer. Mol. Cancer Res. 2019, 17, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Kwon, J.; Woo, J.H.; Kim, K.H.; Lee, K.A. Bcl2l10 mediates the proliferation, invasion and migration of ovarian cancer cells. Int. J. Oncol. 2020, 56, 618–629. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kwon, J.; Lee, K.A. Bcl2l10 induces metabolic alterations in ovarian cancer cells by regulating the TCA cycle enzymes SDHD and IDH1. Oncol. Rep. 2021, 45, 47. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Millar, A.H. Succinate dehydrogenase: The complex roles of a simple enzyme. Curr. Opin. Plant Biol. 2013, 16, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Zhang, H.; Wang, X.; Zhang, X.; Nie, K. The Role of Succinic Acid Metabolism in Ovarian Cancer. Front. Oncol. 2021, 11, 769196. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Zhang, S.; Zhou, J.; Wang, Y.; Di, W. Succinate dehydrogenase subunit B inhibits the AMPK-HIF-1alpha pathway in human ovarian cancer in vitro. J. Ovarian Res. 2014, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspuria, P.P.; Lunt, S.Y.; Varemo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 21. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE 2014, 9, e115708. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Odia, Y.; Orr, B.A.; Bell, W.R.; Eberhart, C.G.; Rodriguez, F.J. cMYC expression in infiltrating gliomas: Associations with IDH1 mutations, clinicopathologic features and outcome. J. Neurooncol. 2013, 115, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Li, S.T.; Huang, D.; Shen, S.; Cai, Y.; Xing, S.; Wu, G.; Jiang, Z.; Hao, Y.; Yuan, M.; Wang, N.; et al. Myc-mediated SDHA acetylation triggers epigenetic regulation of gene expression and tumorigenesis. Nat. Metab. 2020, 2, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Boidot, R.; Vegran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Salem, A.F.; Howell, A.; Sartini, M.; Sotgia, F.; Lisanti, M.P. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1alpha, autophagy and ketone body production. Cell Cycle 2012, 11, 4167–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 603837. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef]
- Lu, J.; Chen, M.; Tao, Z.; Gao, S.; Li, Y.; Cao, Y.; Lu, C.; Zou, X. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017, 8, 76458–76467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Xu, Y.; Wang, F.; Shen, Z.; Tuo, X.; Qian, H.; Wang, H.; Wang, K. Clinical associations between ASCT2 and pmTOR in the pathogenesis and prognosis of epithelial ovarian cancer. Oncol. Rep. 2018, 40, 3725–3733. [Google Scholar] [CrossRef] [Green Version]
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.W.; Cerione, R.A. Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget 2010, 1, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Sheng, X.; Willson, A.K.; Roque, D.R.; Stine, J.E.; Guo, H.; Jones, H.M.; Zhou, C.; Bae-Jump, V.L. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. Relat. Cancer 2015, 22, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Wang, Y.; Zhang, Z.; Lu, J.; Wu, Z.; Shan, Q.; Sun, C.; Wu, D.; Li, M.; Sheng, N.; et al. High expression of glutamate-ammonia ligase is associated with unfavorable prognosis in patients with ovarian cancer. J. Cell. Biochem. 2018, 119, 6008–6015. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [Green Version]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, D.; Avolio, R.; Parri, M.; Romano, S.; Chiarugi, P.; Matassa, D.S.; Esposito, F. Decreased Levels of GSH Are Associated with Platinum Resistance in High-Grade Serous Ovarian Cancer. Antioxidants 2022, 11, 1544. [Google Scholar] [CrossRef]
- Pompella, A.; Corti, A.; Visvikis, A. Redox Mechanisms in Cisplatin Resistance of Cancer Cells: The Twofold Role of Gamma-Glutamyltransferase 1 (GGT1). Front. Oncol. 2022, 12, 920316. [Google Scholar] [CrossRef]
- Pompella, A.; De Tata, V.; Paolicchi, A.; Zunino, F. Expression of gamma-glutamyltransferase in cancer cells and its significance in drug resistance. Biochem. Pharmacol. 2006, 71, 231–238. [Google Scholar] [CrossRef]
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 2016, 7, 41637–41649. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hong, S.; Yang, J.; Zhang, X.; Wang, Y.; Wang, H.; Peng, J.; Hong, L. Targeting purine metabolism in ovarian cancer. J. Ovarian Res. 2022, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Pollari, S.; Kakonen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Riscal, R.; Schrepfer, E.; Arena, G.; Cisse, M.Y.; Bellvert, F.; Heuillet, M.; Rambow, F.; Bonneil, E.; Sabourdy, F.; Vincent, C.; et al. Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol. Cell 2016, 62, 890–902. [Google Scholar] [CrossRef] [Green Version]
- Kaira, K.; Nakamura, K.; Hirakawa, T.; Imai, H.; Tominaga, H.; Oriuchi, N.; Nagamori, S.; Kanai, Y.; Tsukamoto, N.; Oyama, T.; et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) expression in patients with ovarian tumors. Am. J. Transl. Res. 2015, 7, 1161–1171. [Google Scholar]
- Van Nyen, T.; Planque, M.; van Wagensveld, L.; Duarte, J.A.G.; Zaal, E.A.; Talebi, A.; Rossi, M.; Korner, P.R.; Rizzotto, L.; Moens, S.; et al. Serine metabolism remodeling after platinum-based chemotherapy identifies vulnerabilities in a subgroup of resistant ovarian cancers. Nat. Commun. 2022, 13, 4578. [Google Scholar] [CrossRef]
- Antonov, A.; Agostini, M.; Morello, M.; Minieri, M.; Melino, G.; Amelio, I. Bioinformatics analysis of the serine and glycine pathway in cancer cells. Oncotarget 2014, 5, 11004–11013. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, F.; An, Y.; Sun, T.; You, Y.; Yang, Q. PHGDH Is Upregulated at Translational Level and Implicated in Platin-Resistant in Ovarian Cancer Cells. Front. Oncol. 2021, 11, 643129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, M.; Jiao, Y.; Lin, B.; Yang, Q. PHGDH Inhibitor CBR-5884 Inhibits Epithelial Ovarian Cancer Progression via ROS/Wnt/beta-Catenin Pathway and Plays a Synergistic Role with PARP Inhibitor Olaparib. Oxid. Med. Cell. Longev. 2022, 2022, 9029544. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Dong, X.; Meng, D.; Zhi, X.; Yuan, L.; Yao, L. PSAT1 Regulated Oxidation-Reduction Balance Affects the Growth and Prognosis of Epithelial Ovarian Cancer. Onco Targets Ther. 2020, 13, 5443–5453. [Google Scholar] [CrossRef]
- Alves, A.; Bassot, A.; Bulteau, A.L.; Pirola, L.; Morio, B. Glycine Metabolism and Its Alterations in Obesity and Metabolic Diseases. Nutrients 2019, 11, 1356. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Yang, Q.; Dogra, S.K.; Wajapeyee, N. Serine hydroxymethyl transferase 1 stimulates pro-oncogenic cytokine expression through sialic acid to promote ovarian cancer tumor growth and progression. Oncogene 2017, 36, 4014–4024. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Zhang, J.; Xu, M.; Chen, F.; Zi, R.; Yue, J.; Zhang, Y.; Chen, N.; Chin, Y.E. Roles of Mitochondrial Serine Hydroxymethyltransferase 2 (SHMT2) in Human Carcinogenesis. J. Cancer 2021, 12, 5888–5894. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Forshell, T.Z.; Rimpi, S.; Kreutzer, C.; Pretsch, W.; Bornkamm, G.W.; Nilsson, J.A. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012, 8, e1002573. [Google Scholar] [CrossRef] [Green Version]
- Abdelmagid, S.A.; Rickard, J.A.; McDonald, W.J.; Thomas, L.N.; Too, C.K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell. Biochem. 2011, 112, 1084–1092. [Google Scholar] [CrossRef]
- Kishikawa, T.; Otsuka, M.; Tan, P.S.; Ohno, M.; Sun, X.; Yoshikawa, T.; Shibata, C.; Takata, A.; Kojima, K.; Takehana, K.; et al. Decreased miR122 in hepatocellular carcinoma leads to chemoresistance with increased arginine. Oncotarget 2015, 6, 8339–8352. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Zhu, X.; Yang, Y.; Du, X.; Song, K.; Zheng, Q.; Zeng, P.; Yao, Q. SLC7A1 Overexpression Is Involved in Energy Metabolism Reprogramming to Induce Tumor Progression in Epithelial Ovarian Cancer and Is Associated with Immune-Infiltrating Cells. J. Oncol. 2022, 2022, 5864826. [Google Scholar] [CrossRef]
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645. [Google Scholar] [CrossRef]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: A method for identifying cancers sensitive to arginine deprivation. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Feun, L.G.; Marini, A.; Walker, G.; Elgart, G.; Moffat, F.; Rodgers, S.E.; Wu, C.J.; You, M.; Wangpaichitr, M.; Kuo, M.T.; et al. Negative argininosuccinate synthetase expression in melanoma tumours may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br. J. Cancer 2012, 106, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Walts, A.E.; Beach, J.A.; Lester, J.; Bomalaski, J.S.; Walsh, C.S.; Ruprecht Wiedemeyer, W.; Karlan, B.Y.; Orsulic, S. Differential expression of argininosuccinate synthetase in serous and non-serous ovarian carcinomas. J. Pathol. Clin. Res. 2015, 1, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberley, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer 2009, 125, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Erez, A.; Nagamani, S.C.; Shchelochkov, O.A.; Premkumar, M.H.; Campeau, P.M.; Chen, Y.; Garg, H.K.; Li, L.; Mian, A.; Bertin, T.K.; et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011, 17, 1619–1626. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Li, X.; Sun, Y.; Li, L.; Zhang, Q.; Zhu, L.; Zhong, Z.; Wang, M.; Wang, Q.; Liu, Z.; et al. NOS1 expression promotes proliferation and invasion and enhances chemoresistance in ovarian cancer. Oncol. Lett. 2020, 19, 2989–2995. [Google Scholar] [CrossRef] [Green Version]
- Hafliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef] [PubMed]
- Kaji, M.; Kabir-Salmani, M.; Anzai, N.; Jin, C.J.; Akimoto, Y.; Horita, A.; Sakamoto, A.; Kanai, Y.; Sakurai, H.; Iwashita, M. Properties of L-type amino acid transporter 1 in epidermal ovarian cancer. Int. J. Gynecol. Cancer 2010, 20, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Vander Heiden, M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef]
- Hall, T.R.; Wallin, R.; Reinhart, G.D.; Hutson, S.M. Branched chain aminotransferase isoenzymes. Purification and characterization of the rat brain isoenzyme. J. Biol. Chem. 1993, 268, 3092–3098. [Google Scholar] [CrossRef] [PubMed]
- Keita, M.; Wang, Z.Q.; Pelletier, J.F.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Mes-Masson, A.M.; Paquet, E.R.; Bachvarov, D. Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol. Oncol. 2013, 128, 356–363. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Faddaoui, A.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Guillemette, C.; Gobeil, S.; Macdonald, E.; et al. BCAT1 expression associates with ovarian cancer progression: Possible implications in altered disease metabolism. Oncotarget 2015, 6, 31522–31543. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yu, D.; Li, L.; Xiao, J.; Zhu, Y.; Liu, Y.; Mou, L.; Tian, Y.; Chen, L.; Zhu, F.; et al. BCKDK Promotes Ovarian Cancer Proliferation and Migration by Activating the MEK/ERK Signaling Pathway. J. Oncol. 2022, 2022, 3691635. [Google Scholar] [CrossRef]
- Craze, M.L.; Cheung, H.; Jewa, N.; Coimbra, N.D.M.; Soria, D.; El-Ansari, R.; Aleskandarany, M.A.; Wai Cheng, K.; Diez-Rodriguez, M.; Nolan, C.C.; et al. MYC regulation of glutamine-proline regulatory axis is key in luminal B breast cancer. Br. J. Cancer 2018, 118, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.H.; Jung, W.H.; Koo, J.S. Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr. Relat. Cancer 2013, 20, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Jung, W.H.; Koo, J.S. Differential expression of enzymes associated with serine/glycine metabolism in different breast cancer subtypes. PLoS ONE 2014, 9, e101004. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, M.; Bommert, K.S.; Kunz, V.; Kruk, J.; Leich, E.; Rudelius, M.; Bargou, R.; Bommert, K. Glutaminase inhibition in multiple myeloma induces apoptosis via MYC degradation. Oncotarget 2017, 8, 85858–85867. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, Y.; Warden, C.; Chen, S. Cross-talk between ER and HER2 regulates c-MYC-mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2015, 149, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, P.; Zlobec, I.; Nydegger, D.T.; Pujol-Gimenez, J.; Bhardwaj, R.; Shirasawa, S.; Tsunoda, T.; Hediger, M.A. Oncogenic KRAS mutations enhance amino acid uptake by colorectal cancer cells via the hippo signaling effector YAP1. Mol. Oncol. 2021, 15, 2782–2800. [Google Scholar] [CrossRef]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Jiang, L.; Zheng, B.; Gu, W. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J. Biol. Chem. 2015, 290, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, I.; Ciribilli, Y.; Monti, P.; Bisio, A.; Pollegioni, L.; Fronza, G.; Inga, A.; Campomenosi, P. P53 family members modulate the expression of PRODH, but not PRODH2, via intronic p53 response elements. PLoS ONE 2013, 8, e69152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowman, X.H.; Hanse, E.A.; Yang, Y.; Ishak Gabra, M.B.; Tran, T.Q.; Li, H.; Kong, M. p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep. 2019, 26, 3051–3060. [Google Scholar] [CrossRef] [Green Version]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yang, X.; Sun, X.; Xu, X.; Li, X.; Guo, Y.; Wang, J.; Li, X.; Yao, L.; Wang, H.; et al. Effect of PTEN loss on metabolic reprogramming in prostate cancer cells. Oncol. Lett. 2019, 17, 2856–2866. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, C.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.; Meng, Y.; Song, H. CD36: An emerging therapeutic target for cancer and its molecular mechanisms. J. Cancer Res. Clin. Oncol. 2022, 148, 1551–1558. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, A.; Westerberg, R.; Jacobsson, A. Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog. Lipid Res. 2006, 45, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in Cancer Metab.olism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Cardenas, H.; Matei, D. Ovarian Cancer-Why Lipids Matter. Cancers 2019, 11, 1870. [Google Scholar] [CrossRef] [Green Version]
- Nile, A.H.; Hannoush, R.N. Fatty acylation of Wnt proteins. Nat. Chem. Biol. 2016, 12, 60–69. [Google Scholar] [CrossRef]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef] [Green Version]
- Veigel, D.; Wagner, R.; Stubiger, G.; Wuczkowski, M.; Filipits, M.; Horvat, R.; Benhamu, B.; Lopez-Rodriguez, M.L.; Leisser, A.; Valent, P.; et al. Fatty acid synthase is a metabolic marker of cell proliferation rather than malignancy in ovarian cancer and its precursor cells. Int. J. Cancer 2015, 136, 2078–2090. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [Green Version]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [Green Version]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, H.; Yung, M.M.; Chen, F.; Chan, W.S.; Chan, Y.S.; Tsui, S.K.; Ngan, H.Y.; Chan, K.K.; Chan, D.W. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics 2022, 12, 3534–3552. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, F.; Pellegrini, I.; Castagnoli, L.; Vingiani, A.; Lobefaro, R.; Zattarin, E.; Santamaria, M.; Pupa, S.M.; Pruneri, G.; de Braud, F.; et al. Targeting lipid metabolism is an emerging strategy to enhance the efficacy of anti-HER2 therapies in HER2-positive breast cancer. Cancer Lett. 2021, 511, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Oza, B.P.; Ropero, S.; Colomer, R.; Lupu, R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10715–10720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Sun, L.; Li, X.; Wang, J.; Zhu, Y.; Jia, Y.; Tong, Z. SCD5 expression correlates with prognosis and response to neoadjuvant chemotherapy in breast cancer. Sci. Rep. 2021, 11, 8976. [Google Scholar] [CrossRef]
- Jia, J.; Che, L.; Cigliano, A.; Wang, X.; Peitta, G.; Tao, J.; Zhong, S.; Ribback, S.; Evert, M.; Chen, X.; et al. Pivotal Role of Fatty Acid Synthase in c-MYC Driven Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 8467. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef] [Green Version]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [Green Version]
- Bartolacci, C.; Andreani, C.; Vale, G.; Berto, S.; Melegari, M.; Crouch, A.C.; Baluya, D.L.; Kemble, G.; Hodges, K.; Starrett, J.; et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 2022, 13, 4327. [Google Scholar] [CrossRef]
- Gouw, A.M.; Eberlin, L.S.; Margulis, K.; Sullivan, D.K.; Toal, G.G.; Tong, L.; Zare, R.N.; Felsher, D.W. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 4300–4305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Yin, X.; Wu, L.; Qin, Q.; Xu, J. MAPK/P53-mediated FASN expression in bone tumors. Oncol. Lett. 2017, 13, 4035–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, K.; Dizin, E.; Ray, H.; Luquain, C.; Lefai, E.; Foufelle, F.; Billaud, M.; Lenoir, G.M.; Venezia, N.D. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J. Biol. Chem. 2006, 281, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; Hartil, K.; Vaitheesvaran, B.; Arrieta-Cruz, I.; Knight, C.M.; Cook, J.R.; Kammoun, H.L.; Febbraio, M.A.; Gutierrez-Juarez, R.; Kurland, I.J.; et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat. Commun. 2014, 5, 5190. [Google Scholar] [CrossRef] [Green Version]
- Conteduca, V.; Scarpi, E.; Farolfi, A.; Brighi, N.; Rossi, L.; Gurioli, G.; Lolli, C.; Schepisi, G.; Bleve, S.; Gianni, C.; et al. Melphalan as a Promising Treatment for BRCA-Related Ovarian Carcinoma. Front. Oncol. 2021, 11, 716467. [Google Scholar] [CrossRef]
- Davis-Perry, S.; Hernandez, E.; Houck, K.L.; Shank, R. Melphalan for the treatment of patients with recurrent epithelial ovarian cancer. Am. J. Clin. Oncol. 2003, 26, 429–433. [Google Scholar] [CrossRef]
- Osher, D.J.; Kushner, Y.B.; Arseneau, J.; Foulkes, W.D. Melphalan as a treatment for BRCA-related ovarian carcinoma: Can you teach an old drug new tricks? J. Clin. Pathol. 2011, 64, 924–926. [Google Scholar] [CrossRef]
- Zub, K.A.; Sousa, M.M.; Sarno, A.; Sharma, A.; Demirovic, A.; Rao, S.; Young, C.; Aas, P.A.; Ericsson, I.; Sundan, A.; et al. Modulation of cell metabolic pathways and oxidative stress signaling contribute to acquired melphalan resistance in multiple myeloma cells. PLoS ONE 2015, 10, e0119857. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, M.A.; Castilla, J.; Alonso, C.; Perez, J.M. Cisplatin biochemical mechanism of action: From cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 2003, 10, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho-Martinez, S.M.; Prieto-Garcia, L.; Prieto, M.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Subcellular targets of cisplatin cytotoxicity: An integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, X.; Fu, J.; Xu, W.; Yuan, J. The Role of Tumour Metabolism in Cisplatin Resistance. Front. Mol. Biosci. 2021, 8, 691795. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J. Natl. Cancer Inst. 2006, 98, 1462–1473. [Google Scholar] [CrossRef]
- Maccio, A.; Madeddu, C. Cisplatin: An old drug with a newfound efficacy—From mechanisms of action to cytotoxicity. Expert Opin. Pharmacother. 2013, 14, 1839–1857. [Google Scholar] [CrossRef]
- Korkmaz, A.; Topal, T.; Oter, S. Pathophysiological aspects of cyclophosphamide and ifosfamide induced hemorrhagic cystitis; implication of reactive oxygen and nitrogen species as well as PARP activation. Cell Biol. Toxicol. 2007, 23, 303–312. [Google Scholar] [CrossRef]
- Mills, K.A.; Chess-Williams, R.; McDermott, C. Novel insights into the mechanism of cyclophosphamide-induced bladder toxicity: Chloroacetaldehyde’s contribution to urothelial dysfunction in vitro. Arch. Toxicol. 2019, 93, 3291–3303. [Google Scholar] [CrossRef]
- Shi, H.; Hou, B.; Li, H.; Zhou, H.; Du, B. Cyclophosphamide Induces the Ferroptosis of Tumor Cells Through Heme Oxygenase-1. Front. Pharmacol. 2022, 13, 839464. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Porteiro, B.; Fondevila, M.F.; Buque, X.; Gonzalez-Rellan, M.J.; Fernandez, U.; Mora, A.; Beiroa, D.; Senra, A.; Gallego, R.; Ferno, J.; et al. Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Mol. Metab. 2018, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Lopez, M.C.; Soler, F.; Pascual-Figal, D.; Fernandez-Belda, F.; Lax, A. Doxorubicin-induced oxidative stress: The protective effect of nicorandil on HL-1 cardiomyocytes. PLoS ONE 2017, 12, e0172803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22 (Suppl. 11), 3–10. [Google Scholar]
- Yang, Y.; Liu, H.; Li, Z.; Zhao, Z.; Yip-Schneider, M.; Fan, Q.; Schmidt, C.M.; Chiorean, E.G.; Xie, J.; Cheng, L.; et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int. J. Biochem. Mol. Biol. 2011, 2, 89–98. [Google Scholar]
- Gebregiworgis, T.; Bhinderwala, F.; Purohit, V.; Chaika, N.V.; Singh, P.K.; Powers, R. Insights into gemcitabine resistance and the potential for therapeutic monitoring. Metabolomics 2018, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Quinn, M.; Plebanski, M. Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer. Biomed. Res. Int. 2015, 2015, 413076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass-Marmor, L.; Beitner, R. Taxol (paclitaxel) induces a detachment of phosphofructokinase from cytoskeleton of melanoma cells and decreases the levels of glucose 1,6-bisphosphate, fructose 1,6-bisphosphate and ATP. Eur. J. Pharmacol. 1999, 370, 195–199. [Google Scholar] [CrossRef]
- Kollmannsberger, C.; Mross, K.; Jakob, A.; Kanz, L.; Bokemeyer, C. Topotecan—A novel topoisomerase I inhibitor: Pharmacology and clinical experience. Oncology 1999, 56, 1–12. [Google Scholar] [CrossRef]
- Yu, X.W.; Wei, D.; Gao, Y.S.; Du, H.Z.; Yu, B.Y.; Li, R.M.; Qian, C.M.; Luo, X.J.; Yuan, S.T.; Wang, J.S.; et al. Synergistic combination of DT-13 and Topotecan inhibits aerobic glycolysis in human gastric carcinoma BGC-823 cells via NM IIA/EGFR/HK II axis. J. Cell. Mol. Med. 2019, 23, 6622–6634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.; Singh, H. Bevacizumab and ovarian cancer. Ther. Adv. Med. Oncol. 2013, 5, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Curtarello, M.; Tognon, M.; Venturoli, C.; Silic-Benussi, M.; Grassi, A.; Verza, M.; Minuzzo, S.; Pinazza, M.; Brillo, V.; Tosi, G.; et al. Rewiring of Lipid Metabolism and Storage in Ovarian Cancer Cells after Anti-VEGF Therapy. Cells 2019, 8, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponte, J.F.; Ab, O.; Lanieri, L.; Lee, J.; Coccia, J.; Bartle, L.M.; Themeles, M.; Zhou, Y.; Pinkas, J.; Ruiz-Soto, R. Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, Potentiates the Activity of Standard of Care Therapeutics in Ovarian Cancer Models. Neoplasia 2016, 18, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Cantley, L.C. Toward a better understanding of folate metabolism in health and disease. J. Exp. Med. 2019, 216, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Goulooze, S.C.; Cohen, A.F.; Rissmann, R. Olaparib. Br. J. Clin. Pharmacol. 2016, 81, 171–173. [Google Scholar] [CrossRef] [Green Version]
- Berardi, D.; Hunter, Y.; van den Driest, L.; Farrell, G.; Rattray, N.J.W.; Rattray, Z. The Differential Metabolic Signature of Breast Cancer Cellular Response to Olaparib Treatment. Cancers 2022, 14, 3661. [Google Scholar] [CrossRef]
- Moore, K.N.; Mirza, M.R.; Matulonis, U.A. The poly (ADP ribose) polymerase inhibitor niraparib: Management of toxicities. Gynecol. Oncol. 2018, 149, 214–220. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Rais, R.; Poklepovic, A.; Dent, P. Valproate augments Niraparib killing of tumor cells. Cancer Biol. Ther. 2018, 19, 797–808. [Google Scholar] [CrossRef]
- Maccio, A.; Madeddu, C. The mechanism of cancer cell death by PARP inhibitors goes beyond DNA damage alone. Int. J. Cancer 2019, 145, 2594–2596. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Meyer, D.; Sagwal, S.K.; Weltmann, K.D.; von Woedtke, T.; Bekeschus, S. The amino acid metabolism is essential for evading physical plasma-induced tumour cell death. Br. J. Cancer 2021, 124, 1854–1863. [Google Scholar] [CrossRef]
- Arora, S.; Narayan, P.; Ison, G.; Berman, T.; Suzman, D.L.; Wedam, S.; Prowell, T.M.; Ghosh, S.; Philip, R.; Osgood, C.L.; et al. U.S. FDA Drug Approvals for Gynecological Malignancies: A Decade in Review. Clin. Cancer Res. 2022, 28, 1058–1071. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, 133247. [Google Scholar] [CrossRef]







| Class of the Gene | Frequency of Somatic Mutations | Frequency of Germline Mutations | Gene | Genetic Alterations | References |
|---|---|---|---|---|---|
| Oncogenes | 0.9% | Her-2/neu | Amplification | [22,23,24] | |
| c-MYC | Amplification Copy Number Variation | [25,26,27] | |||
| 0.6% | KRAS | Mutations in codon 12 and 13 | [28,29,30] | ||
| BTAK | Amplification | [31,32] | |||
| Tumor Suppressor Genes | 95.9% | TP53 | Loss of Heterozygosity Driver Mutations Missense Mutations | [33,34,35,36,37,38] | |
| 3.5% | 8.2% | BRCA1 | Germline and Somatic mutations | [36,37,38] | |
| 3.2% | 7.9% | BRCA2 | Germline and Somatic mutations | [39,40,41] | |
| 0.6% | PTEN | Driver Mutations | [39,40,41,42,43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murali, R.; Balasubramaniam, V.; Srinivas, S.; Sundaram, S.; Venkatraman, G.; Warrier, S.; Dharmarajan, A.; Gandhirajan, R.K. Deregulated Metabolic Pathways in Ovarian Cancer: Cause and Consequence. Metabolites 2023, 13, 560. https://doi.org/10.3390/metabo13040560
Murali R, Balasubramaniam V, Srinivas S, Sundaram S, Venkatraman G, Warrier S, Dharmarajan A, Gandhirajan RK. Deregulated Metabolic Pathways in Ovarian Cancer: Cause and Consequence. Metabolites. 2023; 13(4):560. https://doi.org/10.3390/metabo13040560
Chicago/Turabian StyleMurali, Roopak, Vaishnavi Balasubramaniam, Satish Srinivas, Sandhya Sundaram, Ganesh Venkatraman, Sudha Warrier, Arun Dharmarajan, and Rajesh Kumar Gandhirajan. 2023. "Deregulated Metabolic Pathways in Ovarian Cancer: Cause and Consequence" Metabolites 13, no. 4: 560. https://doi.org/10.3390/metabo13040560
APA StyleMurali, R., Balasubramaniam, V., Srinivas, S., Sundaram, S., Venkatraman, G., Warrier, S., Dharmarajan, A., & Gandhirajan, R. K. (2023). Deregulated Metabolic Pathways in Ovarian Cancer: Cause and Consequence. Metabolites, 13(4), 560. https://doi.org/10.3390/metabo13040560