

Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb

 , , , and
, , , and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Impact of Maternal Obesity on Maternal Health
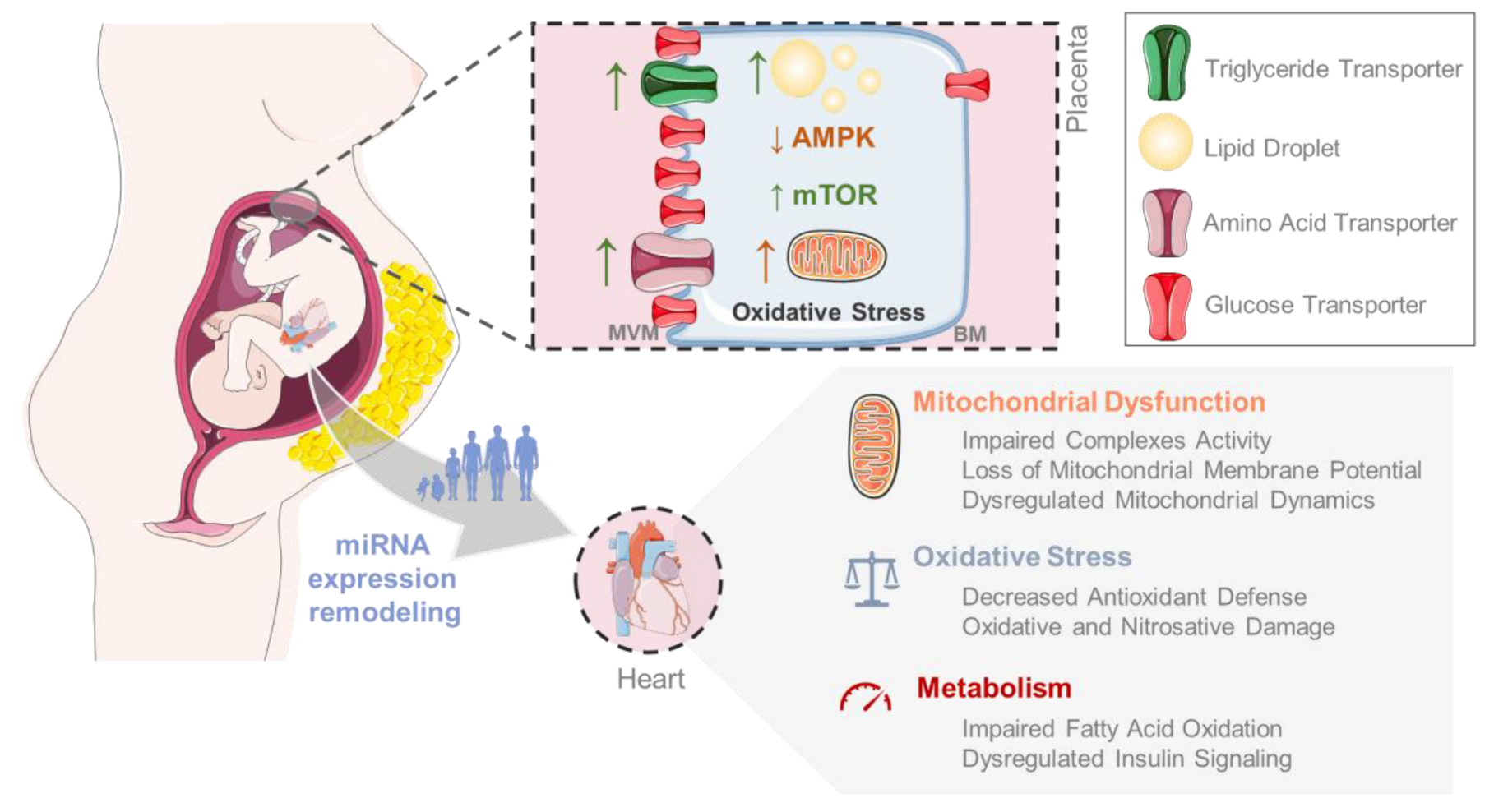
3. Maternal Obesity-Derived Effects in the Fetoplacental Unit
4. Fetal Programming: Womb-Dictated Disease
4.1. Maternal Obesity Impacts Heart Formation and Offspring Cardiac Physiology
4.2. Epigenetic Disruption in Maternal Obesity Progeny
4.3. Sexual Dimorphism in Fetal Programming
4.3.1. The Role of Epigenetic Mechanisms in Sexual Dimorphism
4.3.2. Sex Steroid Hormones
5. The Metabolic Legacy of an Obesogenic Womb
5.1. Maternal Obesity Modulates the Offspring’s Cardiac Insulin Signaling Pathway
5.2. Dysregulation of Glucose and Fatty Acid Oxidation in the MO Offspring
5.3. The Missing Link: Exploring the Role of Mitochondrial Dysfunction in the Development of MO-Induced Cardiovascular Disease in the Offspring
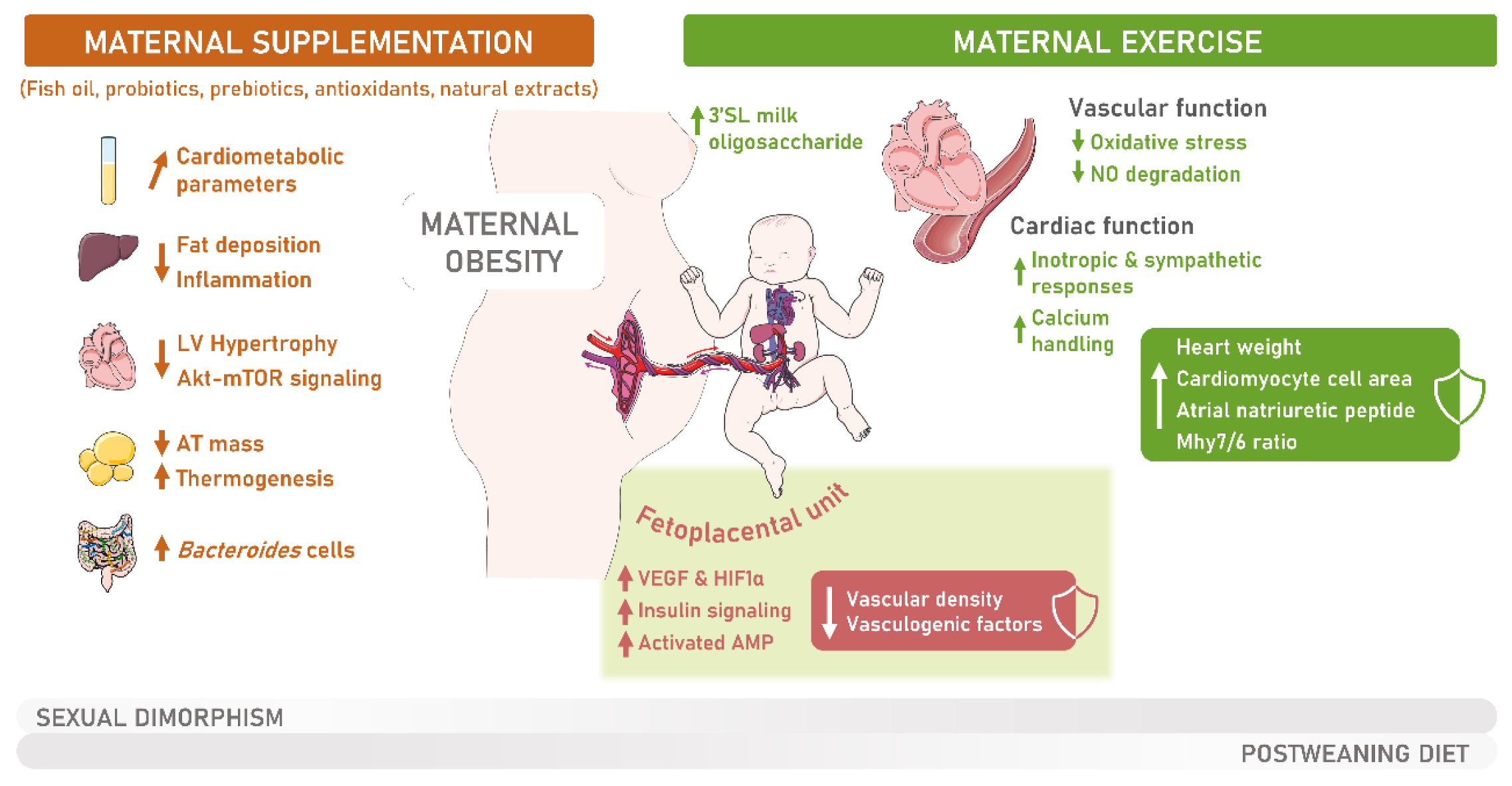
6. Intervention Strategies for Addressing the Metabolic Derangements Caused by Maternal Obesity
6.1. The Influence of Maternal Physical Exercise during Gestation on Offspring’s Overall Health
6.2. Enhancing MO Offspring Cardiovascular Health through Maternal Physical Exercise
7. Final Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Dutton, H.; Borengasser, S.J.; Gaudet, L.M.; Barbour, L.A.; Keely, E.J. Obesity in Pregnancy: Optimizing Outcomes for Mom and Baby. Med. Clin. N. Am. 2018, 102, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Caleyachetty, R.; Cnattingius, S.; Corvalán, C.; Uauy, R.; Herring, S.; Gillman, M.W. Preconceptional and Maternal Obesity: Epidemiology and Health Consequences. Lancet Diabetes Endocrinol. 2016, 4, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, N.; Liu, Y. Association between Maternal Factors and Risk of Congenital Heart Disease in Offspring: A Systematic Review and Meta-Analysis. Matern. Child Health J. 2023, 27, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Angell, S.Y.; McConnell, M.V.; Anderson, C.A.M.; Bibbins-Domingo, K.; Boyle, D.S.; Capewell, S.; Ezzati, M.; de Ferranti, S.; Gaskin, D.J.; Goetzel, R.Z.; et al. The American Heart Association 2030 Impact Goal: A Presidential Advisory From the American Heart Association. Circulation 2020, 141, e120–e138. [Google Scholar] [CrossRef]
- Catalano, P.M.; Shankar, K. Obesity and Pregnancy: Mechanisms of Short Term and Long Term Adverse Consequences for Mother and Child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef]
- Persson, M.; Razaz, N.; Edstedt Bonamy, A.-K.; Villamor, E.; Cnattingius, S. Maternal Overweight and Obesity and Risk of Congenital Heart Defects. J. Am. Coll. Cardiol. 2019, 73, 44–53. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Chu, S.Y.; Kim, S.Y.; Schmid, C.H.; Lau, J. Maternal Obesity and Risk of Neural Tube Defects: A Metaanalysis. Am. J. Obstet. Gynecol. 2008, 198, 611–619. [Google Scholar] [CrossRef]
- Diniz, M.S.; Tocantins, C.; Grilo, L.F.; Pereira, S.P. The Bitter Side of Sugar Consumption: A Mitochondrial Perspective on Obstet Development. Diabetology 2022, 3, 583–595. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of Maternal Obesity on the Long-Term Health of Offspring. Lancet Diabetes Endocrinol. 2017, 5, 53. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, R.; Steegers, E.A.P.; Franco, O.H.; Hofman, A.; Jaddoe, V.W.V. Maternal Weight Gain in Different Periods of Pregnancy and Childhood Cardio-Metabolic Outcomes. The Generation R Study. Int. J. Obes. 2015, 39, 677–685. [Google Scholar] [CrossRef]
- Mamun, A.A.; O’Callaghan, M.; Callaway, L.; Williams, G.; Najman, J.; Lawlor, D.A. Associations of Gestational Weight Gain with Offspring Body Mass Index and Blood Pressure at 21 Years of Ageevidence from a Birth Cohort Study. Circulation 2009, 119, 1720–1727. [Google Scholar] [CrossRef]
- den Harink, T.; Roelofs, M.J.M.; Limpens, J.; Painter, R.C.; Roseboom, T.J.; van Deutekom, A.W. Maternal Obesity in Pregnancy and Children’s Cardiac Function and Structure: A Systematic Review and Meta-Analysis of Evidence from Human Studies. PLoS ONE 2022, 17, e0275236. [Google Scholar] [CrossRef]
- Dong, M.; Zheng, Q.; Ford, S.P.; Nathanielsz, P.W.; Ren, J. Maternal Obesity, Lipotoxicity and Cardiovascular Diseases in Offspring. J. Mol. Cell Cardiol. 2013, 55, 111–116. [Google Scholar] [CrossRef]
- Keleher, M.R.; Zaidi, R.; Shah, S.; Oakley, M.E.; Pavlatos, C.; El Idrissi, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. Maternal High-Fat Diet Associated with Altered Gene Expression, DNA Methylation, and Obesity Risk in Mouse Offspring. PLoS ONE 2018, 13, e0192606. [Google Scholar] [CrossRef] [Green Version]
- Grilo, L.F.; Diniz, M.S.; Tocantins, C.; Areia, A.L.; Pereira, S.P. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities 2022, 2, 236–255. [Google Scholar] [CrossRef]
- George, M.G.; Tong, X.; Bowman, B.A. Prevalence of Cardiovascular Risk Factors and Strokes in Younger Adults. JAMA Neurol. 2017, 74, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Tanvig, M.; Wehberg, S.; Vinter, C.; Joergensen, J.; Ovesen, P.; Beck-Nielsen, H.; Jensen, D.; Christesen, H. Pregestational Body Mass Index Is Related to Neonatal Abdominal Circumference at Birth-a Danish Population-Based Study. BJOG 2013, 120, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Razaz, N.; Villamor, E.; Muraca, G.M.; Bonamy, A.-K.E.; Cnattingius, S. Maternal Obesity and Risk of Cardiovascular Diseases in Offspring: A Population-Based Cohort and Sibling-Controlled Study. Lancet Diabetes Endocrinol. 2020, 8, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Kankowski, L.; Ardissino, M.; McCracken, C.; Lewandowski, A.J.; Leeson, P.; Neubauer, S.; Harvey, N.C.; Petersen, S.E.; Raisi-Estabragh, Z. The Impact of Maternal Obesity on Offspring Cardiovascular Health: A Systematic Literature Review. Front. Endocrinol. 2022, 13, 868441. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Allan, K.M.; Raja, E.A.; Bhattacharya, S.; McNeill, G.; Hannaford, P.C.; Sarwar, N.; Lee, A.J.; Bhattacharya, S.; Norman, J.E. Maternal Obesity during Pregnancy and Premature Mortality from Cardiovascular Event in Adult Offspring: Follow-up of 1 323 275 Person Years. BMJ 2013, 347, f4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Investig. 2021, 51, e13625. [Google Scholar] [CrossRef]
- Ramlakhan, K.P.; Johnson, M.R.; Roos-Hesselink, J.W. Pregnancy and Cardiovascular Disease. Nat. Rev. Cardiol. 2020, 17, 718–731. [Google Scholar] [CrossRef]
- Angueira, A.R.; Ludvik, A.E.; Reddy, T.E.; Wicksteed, B.; Lowe, W.L.; Layden, B.T. New Insights into Gestational Glucose Metabolism: Lessons Learned from 21st Century Approaches. Diabetes 2015, 64, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Catalano, P.M.; Presley, L.; Minium, J.; Mouzon, S.H. De Fetuses of Obese Mothers Develop Insulin Resistance in Utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Newbern, D.; Freemark, M. Placental Hormones and the Control of Maternal Metabolism and Fetal Growth. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 409–416. [Google Scholar] [CrossRef]
- Tuersunjiang, N.; Odhiambo, J.F.; Long, N.M.; Shasa, D.R.; Nathanielsz, P.W.; Ford, S.P. Diet Reduction to Requirements in Obese/Overfed Ewes from Early Gestation Prevents Glucose/Insulin Dysregulation and Returns Fetal Adiposity and Organ Development to Control Levels. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E868–E878. [Google Scholar] [CrossRef] [PubMed]
- Marcinkevage, J.A.; Narayan, K.M.V. Gestational Diabetes Mellitus: Taking It to Heart. Prim Care Obstet. 2011, 5, 81–88. [Google Scholar] [CrossRef]
- Tinius, R.A.; Blankenship, M.M.; Furgal, K.E.; Cade, W.T.; Pearson, K.J.; Rowland, N.S.; Pearson, R.C.; Hoover, D.L.; Maples, J.M. Metabolic Flexibility Is Impaired in Women Who Are Pregnant and Overweight/Obese and Related to Insulin Resistance and Inflammation. Metabolism 2020, 104, 154142. [Google Scholar] [CrossRef] [PubMed]
- Tocantins, C.; Diniz, M.S.; Grilo, L.F.; Pereira, S.P. The Birth of Cardiac Disease: Mechanisms Linking Gestational Obstets Mellitus and Early Onset of Cardiovascular Disease in Offspring. WIREs Mech. Dis. 2022, 14, e1555. [Google Scholar] [CrossRef] [PubMed]
- Mouzon, S.H.; Lassance, L. Endocrine and Metabolic Adaptations to Pregnancy; Impact of Obesity. Horm. Mol. Biol. Clin. Investig. 2015, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lain, K.Y.; Catalano, P.M. Metabolic Changes in Pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef]
- Poston, L.; Harthoorn, L.F.; Van Der Beek, E.M. Obesity in Pregnancy: Implications for the Mother and Lifelong Health of the Child. A Consensus Statement. Pediatr. Res. 2011, 69, 175–180. [Google Scholar] [CrossRef]
- Houttu, N.; Mokkala, K.; Laitinen, K. Overweight and Obesity Status in Pregnant Women Are Related to Intestinal Microbiota and Serum Metabolic and Inflammatory Profiles. Clin. Nutr. 2018, 37, 1955–1966. [Google Scholar] [CrossRef]
- Taylor, K.; Ferreira, D.L.S.; West, J.; Yang, T.; Caputo, M.; Lawlor, D.A. Differences in Pregnancy Metabolic Profiles and Their Determinants between White European and South Asian Women: Findings from the Born in Bradford Cohort. Metabolites 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Kivelä, J.; Sormunen-Harju, H.; Girchenko, P.V.; Huvinen, E.; Stach-Lempinen, B.; Kajantie, E.; Villa, P.M.; Reynolds, R.M.; Hämäläinen, E.K.; Lahti-Pulkkinen, M.; et al. Longitudinal Metabolic Profiling of Maternal Obesity, Gestational Diabetes, and Hypertensive Pregnancy Disorders. J. Clin. Endocrinol. Metab. 2021, 106, e4372. [Google Scholar] [CrossRef]
- Ritchie, S.C.; Würtz, P.; Nath, A.P.; Abraham, G.; Havulinna, A.S.; Fearnley, L.G.; Sarin, A.P.; Kangas, A.J.; Soininen, P.; Aalto, K.; et al. The Biomarker GlycA Is Associated with Chronic Inflammation and Predicts Long-Term Risk of Severe Infection. Cell Syst. 2015, 1, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Newgard, C.B. Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, S.C.; Hunter, S.; Boys, R.J.; Dunlop, W. Serial Study of Factors Influencing Changes in Cardiac Output during Human Pregnancy. Am. J. Physiol. 1989, 256, H1060–H1065. [Google Scholar] [CrossRef] [PubMed]
- Meah, V.L.; Cockcroft, J.R.; Backx, K.; Shave, R.; Stöhr, E.J. Cardiac Output and Related Haemodynamics during Pregnancy: A Series of Meta-Analyses. Heart 2016, 102, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Duvekot, J.J.; Peeters, L.L.H. Maternal Cardiovascular Hemodynamic Adaptation to Pregnancy. Obstet. Gynecol. Surv. 1994, 49, S1–S14. [Google Scholar] [CrossRef]
- De Haas, S.; Ghossein-Doha, C.; Geerts, L.; van Kuijk, S.M.J.; van Drongelen, J.; Spaanderman, M.E.A. Cardiac Remodeling in Normotensive Pregnancy and in Pregnancy Complicated by Hypertension: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2017, 50, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Rodger, M.; Sheppard, D.; Gándara, E.; Tinmouth, A. Haematological Problems in Obstetrics. Best Pr. Res. Clin. Obstet. Gynaecol. 2015, 29, 671–684. [Google Scholar] [CrossRef]
- Hellgren, M.; Blombäck, M. Studies on Blood Coagulation and Fibrinolysis in Pregnancy, during Delivery and in the Puerperium. I. Normal Condition. Gynecol Obstet. Invest 1981, 12, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, P.G.; Macphail, S.; Lind, T. Serial Hematologic Changes and Pregnancy Outcome. Obstet. Gynecol. 1996, 88, 33–39. [Google Scholar] [CrossRef]
- Peck, T.M.; Arias, F. Hematologic Changes Associated with Pregnancy. Clin. Obstet. Gynecol. 1979, 22, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Duvekot, J.J.; Cheriex, E.C.; Pieters, F.A.A.; Menheere, P.P.C.A.; Peeters, L.L.H. Early Pregnancy Changes in Hemodynamics and Volume Homeostasis Are Consecutive Adjustments Triggered by a Primary Fall in Systemic Vascular Tone. Am. J. Obstet. Gynecol. 1993, 169, 1382–1392. [Google Scholar] [CrossRef]
- Ochsenbein-Kölble, N.; Roos, M.; Gasser, T.; Huch, R.; Huch, A.; Zimmermann, R. Cross Sectional Study of Automated Blood Pressure Measurements throughout Pregnancy. BJOG 2004, 111, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Blanco, A.L.Y.; Díaz-López, K.M.; Vilchis-Gil, J.; Diaz-Garcia, H.; Gomez-Lopez, J.; Medina-Bravo, P.; Granados-Riveron, J.T.; Gallardo, J.M.; Klünder-Klünder, M.; Sánchez-Urbina, R. Diet and Maternal Obesity Are Associated with Increased Oxidative Stress in Newborns: A Cross-Sectional Study. Nutrients 2022, 14, 746. [Google Scholar] [CrossRef] [PubMed]
- Stewart, F.M.; Freeman, D.J.; Ramsay, J.E.; Greer, I.A.; Caslake, M.; Ferrell, W.R. Longitudinal Assessment of Maternal Endothelial Function and Markers of Inflammation and Placental Function throughout Pregnancy in Lean and Obese Mothers. J. Clin. Endocrinol. Metab. 2007, 92, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, J.E.; Ferrell, W.R.; Crawford, L.; Wallace, A.M.; Greer, I.A.; Sattar, N. Maternal Obesity Is Associated with Dysregulation of Metabolic, Vascular, and Inflammatory Pathways. J. Clin. Endocrinol. Metab. 2002, 87, 4231–4237. [Google Scholar] [CrossRef]
- Contreras-Duarte, S.; Carvajal, L.; Garchitorena, M.J.; Subiabre, M.; Fuenzalida, B.; Cantin, C.; Farías, M.; Leiva, A. Gestational Diabetes Mellitus Treatment Schemes Modify Maternal Plasma Cholesterol Levels Dependent to Women’s Weight: Possible Impact on Feto-Placental Vascular Function. Nutrients 2020, 12, 506. [Google Scholar] [CrossRef] [Green Version]
- Gallos, I.; Sivakumar, K.; Kilby, M.; Coomarasamy, A.; Thangaratinam, S.; Vatish, M. Pre-Eclampsia Is Associated with, and Preceded by, Hypertriglyceridaemia: A Meta-Analysis. BJOG 2013, 120, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.; Diamond, P.; Singh, G.; Bell, C. Brief Overview of Maternal Triglycerides as a Risk Factor for Pre-Eclampsia. BJOG 2006, 113, 379–386. [Google Scholar] [CrossRef]
- Melchor, I.; Burgos, J.; Del Campo, A.; Aiartzaguena, A.; Gutiérrez, J.; Melchor, J.C. Effect of Maternal Obesity on Pregnancy Outcomes in Women Delivering Singleton Babies: A Historical Cohort Study. J. Perinat. Med. 2019, 47, 625–630. [Google Scholar] [CrossRef]
- Odutayo, A.; Hladunewich, M. Obstetric Nephrology: Renal Hemodynamic and Metabolic Physiology in Normal Pregnancy. Clin. J. Am. Soc. Nephrol. 2012, 7, 2073–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Haas, S.; Ghossein-Doha, C.; van Kuijk, S.M.J.; van Drongelen, J.; Spaanderman, M.E.A. Physiological Adaptation of Maternal Plasma Volume during Pregnancy: A Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2017, 49, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozaki, A.; Matsuda, Y.; Satoh, S.; Saito, S. Comparison of Risk Factors for Gestational Hypertension and Preeclampsia in Japanese Singleton Pregnancies. J. Obstet. Gynaecol. Res. 2013, 39, 492–499. [Google Scholar] [CrossRef]
- Steegers, E.A.P.; Von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-Eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Hylenius, S.; Andersen, A.M.N.; Melbye, M.; Hviid, T.V.F. Association between HLA-G Genotype and Risk of Pre-Eclampsia: A Case-Control Study Using Family Triads. Mol. Hum. Reprod. 2004, 10, 237–246. [Google Scholar] [CrossRef] [Green Version]
- KHONG, T.Y.; DE WOLF, F.; ROBERTSON, W.B.; BROSENS, I. Inadequate Maternal Vascular Response to Placentation in Pregnancies Complicated by Pre-Eclampsia and by Small-for-Gestational Age Infants. Br. J. Obstet. Gynaecol. 1986, 93, 1049–1059. [Google Scholar] [CrossRef]
- Kelly, A.C.; Powell, T.L.; Jansson, T. Placental Function in Maternal Obesity. Clin. Sci. 2020, 134, 961. [Google Scholar] [CrossRef]
- Johns, E.C.; Denison, F.C.; Reynolds, R.M. The Impact of Maternal Obesity in Pregnancy on Placental Glucocorticoid and Macronutrient Transport and Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boss, A.L.; Chamley, L.W.; James, J.L. Placental Formation in Early Pregnancy: How Is the Centre of the Placenta Made? Hum. Reprod. Update 2018, 24, 750–760. [Google Scholar] [CrossRef] [PubMed]
- GJ, B. The Fine Structure of the Human Placental Villus as Revealed by Scanning Electron Microscopy. Scanning Microsc. 1987, 1, 1811–1828. [Google Scholar]
- Burton, G.J.; Fowden, A.L. The Placenta: A Multifaceted, Transient Organ. Philos. Trans. R Soc. Lond B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C.P. Rheological and Physiological Consequences of Conversion of the Maternal Spiral Arteries for Uteroplacental Blood Flow during Human Pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollheimer, J.; Vondra, S.; Baltayeva, J.; Beristain, A.G.; Knöfler, M. Regulation of Placental Extravillous Trophoblasts by the Maternal Uterine Environment. Front. Immunol. 2018, 9, 2597. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, G. Review: Placental Syncytiotrophoblast Membranes—Domains, Subdomains and Microdomains. Placenta 2011, 32 (Suppl. S2), S196–S202. [Google Scholar] [CrossRef]
- Mata-Greenwood, E.; Huber, H.F.; Li, C.; Nathanielsz, P.W. Role of Pregnancy and Obesity on Vitamin D Status, Transport, and Metabolism in Baboons. Am. J. Physiol.-Endocrinol. Metab. 2019, 316, E63–E72. [Google Scholar] [CrossRef] [Green Version]
- Draycott, S.A.V.; Daniel, Z.; Khan, R.; Muhlhausler, B.S.; Elmes, M.J.; Langley-Evans, S.C. Expression of Cholesterol Packaging and Transport Genes in Human and Rat Placenta: Impact of Obesity and a High-Fat Diet. J. Dev. Orig. Health Dis. 2020, 11, 222–227. [Google Scholar] [CrossRef]
- Loardi, C.; Falchetti, M.; Prefumo, F.; Facchetti, F.; Frusca, T. Placental Morphology in Pregnancies Associated with Pregravid Obesity. J. Matern Fetal. Neonatal Med. 2016, 29, 2611–2616. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.A.; Riley, S.C.; Reynolds, R.M.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.N.; Norman, J.E.; Denison, F.C. Placental Structure and Inflammation in Pregnancies Associated with Obesity. Placenta 2011, 32, 247–254. [Google Scholar] [CrossRef]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in Pregnancy Stimulates Macrophage Accumulation and Inflammation in the Placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Stirrat, L.I.; O’Reilly, J.R.; Barr, S.M.; Andrew, R.; Riley, S.C.; Howie, A.F.; Bowman, M.; Smith, R.; Lewis, J.G.; Denison, F.C.; et al. Decreased Maternal Hypothalamic-Pituitary-Adrenal Axis Activity in Very Severely Obese Pregnancy: Associations with Birthweight and Gestation at Delivery. Psychoneuroendocrinology 2016, 63, 135–143. [Google Scholar] [CrossRef]
- Jones, H.N.; Woollett, L.A.; Barbour, N.; Prasad, P.D.; Powell, T.L.; Jansson, T. High-Fat Diet before and during Pregnancy Causes Marked up-Regulation of Placental Nutrient Transport and Fetal Overgrowth in C57/BL6 Mice. FASEB J 2009, 23, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Furesz, T.C.; Laurent, R.S.; Smith, C.H.; Fant, M.E. Spatial Polarization of Insulin-Like Growth Factor Receptors on the Human Syncytiotrophoblast. Pediatr. Res. 1997, 41, 258–265. [Google Scholar] [CrossRef] [Green Version]
- James-Allan, L.B.; Arbet, J.; Teal, S.B.; Powell, T.L.; Jansson, T. Insulin Stimulates GLUT4 Trafficking to the Syncytiotrophoblast Basal Plasma Membrane in the Human Placenta. J. Clin. Endocrinol. Metab. 2019, 104, 4225–4238. [Google Scholar] [CrossRef]
- Ebenbichler, C.F.; Kaser, S.; Laimer, M.; Wolf, H.J.; Patsch, J.R.; Illsley, N.P. Polar Expression and Phosphorylation of Human Leptin Receptor Isoforms in Paired, Syncytial, Microvillous and Basal Membranes from Human Term Placenta. Placenta 2002, 23, 516–521. [Google Scholar] [CrossRef]
- Raubenheimer, P.J.; Young, E.A.; Andrew, R.; Seckl, J.R. The Role of Corticosterone in Human Hypothalamic-Pituitary-Adrenal Axis Feedback. Clin. Endocrinol. 2006, 65, 22–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of Placental MTOR Signaling and Amino Acid Transporters in Obese Women Giving Birth to Large Babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, J.; Muralimanoharan, S.; Maloyan, A.; Myatt, L. Impaired Mitochondrial Function in Human Placenta with Increased Maternal Adiposity. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E419–E425. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.B.; Jansson, T. Novel Roles of Mechanistic Target of Rapamycin Signaling in Regulating Fetal Growth†. Biol. Reprod. 2019, 100, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Mammalian Target of Rapamycin Signalling Modulates Amino Acid Uptake by Regulating Transporter Cell Surface Abundance in Primary Human Trophoblast Cells. J. Physiol. 2013, 591, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Nathanielsz, P.W.; Powell, T.L.; Jansson, T. Maternal Folate Deficiency Causes Inhibition of MTOR Signaling, down-Regulation of Placental Amino Acid Transporters and Fetal Growth Restriction in Mice. Sci. Rep. 2017, 7, 3982. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Powell, T.L.; Jansson, T. MTOR Folate Sensing Links Folate Availability to Trophoblast Cell Function. J. Physiol. 2017, 595, 4189–4206. [Google Scholar] [CrossRef] [Green Version]
- Rosario, F.J.; Gupta, M.B.; Myatt, L.; Powell, T.L.; Glenn, J.P.; Cox, L.; Jansson, T. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci. Rep. 2019, 9, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, R.; Drake, M.; Seckl, J.; O’Reilly, J.R. Effects of Maternal Stress and Obesity on Human Feto-Placental Glucocorticoid Exposure. Ph.D. Dissertation, University of Edinburgh, Edinburgh, UK, 2014. [Google Scholar]
- Martino, J.; Sebert, S.; Segura, M.T.; Garcia-Valdés, L.; Florido, J.; Padilla, M.C.; Marcos, A.; Rueda, R.; McArdle, H.J.; Budge, H.; et al. Maternal Body Weight and Gestational Obstet. Differentially Influence Placental and Pregnancy Outcomes. J. Clin. Endocrinol. Metab. 2016, 101, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Calabuig-Navarro, V.; Haghiac, M.; Minium, J.; Glazebrook, P.; Ranasinghe, G.C.; Hoppel, C.; De-Mouzon, S.H.; Catalano, P.; O’Tierney-Ginn, P. Effect of Maternal Obesity on Placental Lipid Metabolism. Endocrinology 2017, 158, 2543–2555. [Google Scholar] [CrossRef]
- Hastie, R.; Lappas, M. The Effect of Pre-Existing Maternal Obesity and Obstet. on Placental Mitochondrial Content and Electron Transport Chain Activity. Placenta 2014, 35, 673–683. [Google Scholar] [CrossRef]
- Shekhawat, P.; Bennett, M.J.; Sadovsky, Y.; Nelson, D.M.; Rakheja, D.; Strauss, A.W. Human Placenta Metabolizes Fatty Acids: Implications for Fetal Fatty Acid Oxidation Disorders and Maternal Liver Diseases. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1098–E1105. [Google Scholar] [CrossRef] [Green Version]
- Rakheja, D.; Bennett, M.J.; Foster, B.M.; Domiati-Saad, R.; Rogers, B.B. Evidence for Fatty Acid Oxidation in Human Placenta, and the Relationship of Fatty Acid Oxidation Enzyme Activities with Gestational Age. Placenta 2002, 23, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Lager, S.; Powell, T.L. Regulation of Nutrient Transport across the Placenta. J. Pregnancy 2012, 2012, 179827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bucher, M.; Myatt, L. Use of Glucose, Glutamine, and Fatty Acids for Trophoblast Respiration in Lean Women, Women with Obesity, and Women with Gestational Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 4178–4187. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.J.; Ma, Y.; Long, N.M.; Du, M.; Ford, S.P. Maternal Obesity Markedly Increases Placental Fatty Acid Transporter Expression and Fetal Blood Triglycerides at Midgestation in the Ewe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1224–R1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolahi, K.S.; Valent, A.M.; Thornburg, K.L. Cytotrophoblast, Not Syncytiotrophoblast, Dominates Glycolysis and Oxidative Phosphorylation in Human Term Placenta. Sci. Rep. 2017, 7, srep42941. [Google Scholar] [CrossRef] [Green Version]
- Saben, J.; Lindsey, F.; Zhong, Y.; Thakali, K.; Badger, T.M.; Andres, A.; Gomez-Acevedo, H.; Shankar, K. Maternal Obesity Is Associated with a Lipotoxic Placental Environment. Placenta 2014, 35, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Hirschmugl, B.; Desoye, G.; Catalano, P.; Klymiuk, I.; Scharnagl, H.; Payr, S.; Kitzinger, E.; Schliefsteiner, C.; Lang, U.; Wadsack, C.; et al. Maternal Obesity Modulates Intracellular Lipid Turnover in the Human Term Placenta. Int. J. Obes. 2017, 41, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Altmäe, S.; Segura, M.T.; Esteban, F.J.; Bartel, S.; Brandi, P.; Irmler, M.; Beckers, J.; Demmelmair, H.; López-Sabater, C.; Koletzko, B.; et al. Maternal Pre-Pregnancy Obesity Is Associated with Altered Placental Transcriptome. PLoS ONE 2017, 12, e0169223. [Google Scholar] [CrossRef] [Green Version]
- Bildirici, I.; Schaiff, W.T.; Chen, B.; Morizane, M.; Oh, S.Y.; O’Brien, M.; Sonnenberg-Hirche, C.; Chu, T.; Barak, Y.; Nelson, D.M.; et al. PLIN2 Is Essential for Trophoblastic Lipid Droplet Accumulation and Cell Survival during Hypoxia. Endocrinology 2018, 159, 3937–3949. [Google Scholar] [CrossRef] [Green Version]
- Saben, J.; Zhong, Y.; Gomez-Acevedo, H.; Thakali, K.M.; Borengasser, S.J.; Andres, A.; Shankar, K. Early Growth Response Protein-1 Mediates Lipotoxicity-Associated Placental Inflammation: Role in Maternal Obesity. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1. [Google Scholar] [CrossRef]
- Lager, S.; Jansson, T.; Powell, T.L. Differential Regulation of Placental Amino Acid Transport by Saturated and Unsaturated Fatty Acids. Am. J. Physiol. Cell Physiol. 2014, 307, C738–C744. [Google Scholar] [CrossRef] [Green Version]
- Gaccioli, F.; Aye, I.L.M.H.; Roos, S.; Lager, S.; Ramirez, V.I.; Kanai, Y.; Powell, T.L.; Jansson, T. Expression and Functional Characterisation of System L Amino Acid Transporters in the Human Term Placenta. Reprod. Biol. Endocrinol. 2015, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelo, J.S.; Jorge, S.M.; Martinez, F.E. Amino Acid Composition of Parturient Plasma, the Intervillous Space of the Placenta and the Umbilical Vein of Term Newborn Infants. Braz. J. Med. Biol. Res. 2004, 37, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleal, J.K.; Lofthouse, E.M.; Sengers, B.G.; Lewis, R.M. A Systems Perspective on Placental Amino Acid Transport. J. Physiol. 2018, 596, 5511–5522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.A.; Barrett, H.L.; Dekker Nitert, M. Review: Placental Transport and Metabolism of Energy Substrates in Maternal Obesity and Diabetes. Placenta 2017, 54, 59–67. [Google Scholar] [CrossRef] [Green Version]
- von Versen-Höynck, F.; Rajakumar, A.; Parrott, M.S.; Powers, R.W. Leptin Affects System A Amino Acid Transport Activity in the Human Placenta: Evidence for STAT3 Dependent Mechanisms. Placenta 2009, 30, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Tessier, D.R.; Ferraro, Z.M.; Gruslin, A. Role of Leptin in Pregnancy: Consequences of Maternal Obesity. Placenta 2013, 34, 205–211. [Google Scholar] [CrossRef]
- Farley, D.M.; Choi, J.; Dudley, D.J.; Li, C.; Jenkins, S.L.; Myatt, L.; Nathanielsz, P.W. Placental Amino Acid Transport and Placental Leptin Resistance in Pregnancies Complicated by Maternal Obesity. Placenta 2010, 31, 718–724. [Google Scholar] [CrossRef]
- Jansson, N.; Greenwood, S.L.; Johansson, B.R.; Powell, T.L.; Jansson, T. Leptin Stimulates the Activity of the System A Amino Acid Transporter in Human Placental Villous Fragments. J. Clin. Endocrinol. Metab. 2003, 88, 1205–1211. [Google Scholar] [CrossRef]
- White, V.; González, E.; Capobianco, E.; Pustovrh, C.; Martínez, N.; Higa, R.; Baier, M.; Jawerbaum, A. Leptin Modulates Nitric Oxide Production and Lipid Metabolism in Human Placenta. Reprod. Fertil. Dev. 2006, 18, 425–432. [Google Scholar] [CrossRef]
- Cameo, P.; Bischof, P.; Calvo, J.C. Effect of Leptin on Progesterone, Human Chorionic Gonadotropin, and Interleukin-6 Secretion by Human Term Trophoblast Cells in Culture. Biol. Reprod. 2003, 68, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illsley, N.P.; Baumann, M.U. Human Placental Glucose Transport in Fetoplacental Growth and Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165359. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.U.; Deborde, S.; Illsley, N.P. Placental Glucose Transfer and Fetal Growth. Endocrine 2002, 19, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Acosta, O.; Ramirez, V.I.; Lager, S.; Gaccioli, F.; Dudley, D.J.; Powell, T.L.; Jansson, T. Increased Glucose and Placental GLUT-1 in Large Infants of Obese Nondiabetic Mothers. Am. J. Obstet. Gynecol. 2015, 212, 227.e1–227.e7. [Google Scholar] [CrossRef]
- Castillo-Castrejon, M.; Jansson, T.; Powell, T.L. No Evidence of Attenuation of Placental Insulin-Stimulated Akt Phosphorylation and Amino Acid Transport in Maternal Obesity and Gestational Diabetes Mellitus. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E1037–E1049. [Google Scholar] [CrossRef]
- Karl, P.I.; Alpy, K.L.; Fisher, S.E. Amino Acid Transport by the Cultured Human Placental Trophoblast: Effect of Insulin on AIB Transport. Am. J. Physiol. 1992, 262, C834–C839. [Google Scholar] [CrossRef]
- Roos, S.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Regulation of Placental Amino Acid Transporter Activity by Mammalian Target of Rapamycin. Am. J. Physiol. Cell Physiol. 2009, 296, C142–C150. [Google Scholar] [CrossRef] [Green Version]
- Ericsson, A.; Hamark, B.; Jansson, N.; Johansson, B.R.; Powell, T.L.; Jansson, T. Hormonal Regulation of Glucose and System A Amino Acid Transport in First Trimester Placental Villous Fragments. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R656–R662. [Google Scholar] [CrossRef] [Green Version]
- Olausson, H.; Löf, M.; Brismar, K.; Forsum, E.; Sohlström, A. Maternal Serum Concentrations of Insulin-like Growth Factor (IGF)-I and IGF Binding Protein-1 before and during Pregnancy in Relation to Maternal Body Weight and Composition and Infant Birth Weight. Br. J. Nutr. 2010, 104, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.U.; Schneider, H.; Malek, A.; Palta, V.; Surbek, D.V.; Sager, R.; Zamudio, S.; Illsley, N.P. Regulation of Human Trophoblast GLUT1 Glucose Transporter by Insulin-Like Growth Factor I (IGF-I). PLoS ONE 2014, 9, e106037. [Google Scholar] [CrossRef]
- Kniss, D.A.; Shubert, P.J.; Zimmerman, P.D.; Landon, M.B.; Gabbe, S.G. Insulinlike Growth Factors. Their Regulation of Glucose and Amino Acid Transport in Placental Trophoblasts Isolated from First-Trimester Chorionic Villi. J. Reprod. Med. 1994, 39, 249–256. [Google Scholar]
- Bhaumick, B.; George, D.; Bala, R.M. Potentiation of Epidermal Growth Factor-Induced Differentiation of Cultured Human Placental Cells by Insulin-like Growth Factor-I. J. Clin. Endocrinol. Metab. 1992, 74, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Sferruzzi-Perri, A.N.; Sandovici, I.; Constancia, M.; Fowden, A.L. Placental Phenotype and the Insulin-like Growth Factors: Resource Allocation to Fetal Growth. J. Physiol. 2017, 595, 5057–5093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aye, I.L.M.H.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing Maternal Body Mass Index Is Associated with Systemic Inflammation in the Mother and the Activation of Distinct Placental Inflammatory Pathways. Biol. Reprod. 2014, 90, 129. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Jansson, T.; Powell, T.L. TNF-α Stimulates System A Amino Acid Transport in Primary Human Trophoblast Cells Mediated by P38 MAPK Signaling. Physiol. Rep. 2015, 3, e12594. [Google Scholar] [CrossRef]
- Jones, H.N.; Jansson, T.; Powell, T.L. IL-6 Stimulates System A Amino Acid Transporter Activity in Trophoblast Cells through STAT3 and Increased Expression of SNAT2. Am. J. Physiol. Cell Physiol. 2009, 297, C1228–C1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lager, S.; Jansson, N.; Olsson, A.L.; Wennergren, M.; Jansson, T.; Powell, T.L. Effect of IL-6 and TNF-α on Fatty Acid Uptake in Cultured Human Primary Trophoblast Cells. Placenta 2011, 32, 121–127. [Google Scholar] [CrossRef]
- Varastehpour, A.; Radaelli, T.; Minium, J.; Ortega, H.; Herrera, E.; Catalano, P.; Hauguel-de Mouzon, S. Activation of Phospholipase A2 Is Associated with Generation of Placental Lipid Signals and Fetal Obesity. J. Clin. Endocrinol. Metab. 2006, 91, 248–255. [Google Scholar] [CrossRef]
- Jansson, N.; Nilsfelt, A.; Gellerstedt, M.; Wennergren, M.; Rossander-Hulthén, L.; Powell, T.L.; Jansson, T. Maternal Hormones Linking Maternal Body Mass Index and Dietary Intake to Birth Weight. Am. J. Clin. Nutr. 2008, 87, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Hendler, I.; Blackwell, S.C.; Mehta, S.H.; Whitty, J.E.; Russell, E.; Sorokin, Y.; Cotton, D.B. The Levels of Leptin, Adiponectin, and Resistin in Normal Weight, Overweight, and Obese Pregnant Women with and without Preeclampsia. Am. J. Obstet. Gynecol. 2005, 193, 979–983. [Google Scholar] [CrossRef]
- Nien, J.K.; Mazaki-Tovi, S.; Romero, R.; Erez, O.; Kusanovic, J.P.; Gotsch, F.; Pineles, B.L.; Gomez, R.; Edwin, S.; Mazor, M.; et al. Plasma Adiponectin Concentrations in Non Pregnant, Normal Pregnancy and Overweight Pregnant Women. J. Perinat Med. 2007, 35, 522. [Google Scholar] [CrossRef] [PubMed]
- Vernini, J.M.; Moreli, J.B.; Costa, R.A.A.; Negrato, C.A.; Rudge, M.V.C.; Calderon, I.M.P. Maternal Adipokines and Insulin as Biomarkers of Pregnancies Complicated by Overweight and Obesity. Diabetol. Metab. Syndr. 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.N.; Jansson, T.; Powell, T.L. Full-Length Adiponectin Attenuates Insulin Signaling and Inhibits Insulin-Stimulated Amino Acid Transport in Human Primary Trophoblast Cells. Diabetes 2010, 59, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aye, I.L.M.H.; Rosario, F.J.; Powell, T.L.; Jansson, T. Adiponectin Supplementation in Pregnant Mice Prevents the Adverse Effects of Maternal Obesity on Placental Function and Fetal Growth. Proc. Natl. Acad. Sci. USA 2015, 112, 12858–12863. [Google Scholar] [CrossRef] [PubMed]
- Ahlsson, F.; Diderholm, B.; Ewald, U.; Jonsson, B.; Forslund, A.; Stridsberg, M.; Gustafsson, J. Adipokines and Their Relation to Maternal Energy Substrate Production, Insulin Resistance and Fetal Size. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 168, 26–29. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Gao, X.; Weintraub, S.T.; Jansson, T.; Powell, T.L. Adiponectin Inhibits Insulin Function in Primary Trophoblasts by PPARα-Mediated Ceramide Synthesis. Mol. Endocrinol. 2014, 28, 512–524. [Google Scholar] [CrossRef] [Green Version]
- Duval, F.; Dos Santos, E.; Poidatz, D.; Sérazin, V.; Gronier, H.; Vialard, F.; Dieudonné, M.N. Adiponectin Inhibits Nutrient Transporters and Promotes Apoptosis in Human Villous Cytotrophoblasts: Involvement in the Control of Fetal Growth. Biol. Reprod. 2016, 94, 1–12. [Google Scholar] [CrossRef]
- Malti, N.; Merzouk, H.; Merzouk, S.A.; Loukidi, B.; Karaouzene, N.; Malti, A.; Narce, M. Oxidative Stress and Maternal Obesity: Feto-Placental Unit Interaction. Placenta 2014, 35, 411–416. [Google Scholar] [CrossRef]
- Fattuoni, C.; Mandò, C.; Palmas, F.; Anelli, G.M.; Novielli, C.; Parejo Laudicina, E.; Savasi, V.M.; Barberini, L.; Dessì, A.; Pintus, R.; et al. Preliminary Metabolomics Analysis of Placenta in Maternal Obesity. Placenta 2018, 61, 89–95. [Google Scholar] [CrossRef]
- Roberts, V.H.J.; Smith, J.; McLea, S.A.; Heizer, A.B.; Richardson, J.L.; Myatt, L. Effect of Increasing Maternal Body Mass Index on Oxidative and Nitrative Stress in the Human Placenta. Placenta 2009, 30, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Lee, K.J. Post-Translational Modifications and Their Biological Functions: Proteomic Analysis and Systematic Approaches. J. Biochem. Mol. Biol. 2004, 37, 35–44. [Google Scholar] [CrossRef]
- Alcala, M.; Gutierrez-Vega, S.; Castro, E.; Guzman-Gutiérrez, E.; Ramos-Álvarez, M.P.; Viana, M. Antioxidants and Oxidative Stress: Focus in Obese Pregnancies. Front. Physiol. 2018, 9, 1569. [Google Scholar] [CrossRef] [Green Version]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.-L. Oxidative Stress in Placental Pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef]
- Wallace, J.G.; Bellissimo, C.J.; Yeo, E.; Fei Xia, Y.; Petrik, J.J.; Surette, M.G.; Bowdish, D.M.E.; Sloboda, D.M. Obesity during Pregnancy Results in Maternal Intestinal Inflammation, Placental Hypoxia, and Alters Fetal Glucose Metabolism at Mid-Gestation. Sci. Rep. 2019, 9, 17621. [Google Scholar] [CrossRef]
- Louey, S.; Jonker, S.S.; Giraud, G.D.; Thornburg, K.L. Placental Insufficiency Decreases Cell Cycle Activity and Terminal Maturation in Fetal Sheep Cardiomyocytes. J. Physiol. 2007, 580, 639–648. [Google Scholar] [CrossRef]
- Muñoz-Chápuli, R.; Pérez-Pomares, J.M. Cardiogenesis: An Embryological Perspective. J. Cardiovasc. Transl. Res. 2010, 3, 37–48. [Google Scholar] [CrossRef]
- Borasch, K.; Richardson, K.; Plendl, J. Cardiogenesis with a Focus on Vasculogenesis and Angiogenesis. J. Vet. Med. Ser. C Anat. Histol. Embryol. 2020, 49, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [Green Version]
- Lock, M.C.; Tellam, R.L.; Botting, K.J.; Wang, K.C.W.; Selvanayagam, J.B.; Brooks, D.A.; Seed, M.; Morrison, J.L. The Role of MiRNA Regulation in Fetal Cardiomyocytes, Cardiac Maturation and the Risk of Heart Disease in Adults. J. Physiol. 2018, 596, 5625–5640. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.; Cowan, D.B.; Emani, S.M.; del Nido, P.J.; McCully, J.D. Mitochondrial Transplantation in Myocardial Ischemia and Reperfusion Injury. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 982, pp. 595–619. [Google Scholar]
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Li, J.; Umar, S.; Amjedi, M.; Iorga, A.; Sharma, S.; Nadadur, R.; Regitz-Zagrosek, V.; Eghbali, M. New Frontiers in Heart Hypertrophy during Pregnancy. Am. J. Cardiovasc. Dis. 2012, 2, 192–207. [Google Scholar] [PubMed]
- Hou, J.; Kang, Y.J. Regression of Pathological Cardiac Hypertrophy: Signaling Pathways and Therapeutic Targets. Pharmacol. Ther. 2012, 135, 337–354. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, X.; Guo, Z.; Zhong, Y.; Wang, P.; Li, J.; Li, Z.; Liu, P. SIRT6 Suppresses NFATc4 Expression and Activation in Cardiomyocyte Hypertrophy. Front. Pharmacol. 2019, 9, 1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Delgado-Olguin, P. Embryonic Programming of Heart Disease in Response to Obesity during Pregnancy. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165402. [Google Scholar] [CrossRef]
- Kempf, T.; Wollert, K.C. Nitric Oxide and the Enigma of Cardiac Hypertrophy. BioEssays 2004, 26, 608–615. [Google Scholar] [CrossRef]
- Rainer, P.P.; Kass, D.A. Old Dog, New Tricks: Novel Cardiac Targets and Stress Regulation by Protein Kinase G. Cardiovasc. Res. 2016, 111, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Govindsamy, A.; Naidoo, S.; Cerf, M.E. Cardiac Development and Transcription Factors: Insulin Signalling, Insulin Resistance, and Intrauterine Nutritional Programming of Cardiovascular Disease. J. Nutr. Metab. 2018, 2018, 8547976. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Twinn, D.S.; Blackmore, H.L.; Siggens, L.; Giussani, D.A.; Cross, C.M.; Foo, R.; Ozanne, S.E. The Programming of Cardiac Hypertrophy in the Offspring by Maternal Obesity Is Associated with Hyperinsulinemia, AKT, ERK, and MTOR Activation. Endocrinology 2012, 153, 5961–5971. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Lindsley, S.R.; Comstock, S.M.; Takahashi, D.L.; Evans, A.E.; He, G.W.; Thornburg, K.L.; Grove, K.L. Maternal High-Fat Diet Impacts Endothelial Function in Nonhuman Primate Offspring. Int. J. Obes. 2013, 37, 254–262. [Google Scholar] [CrossRef] [Green Version]
- Mdaki, K.S.; Larsen, T.D.; Wachal, A.L.; Schimelpfenig, M.D.; Weaver, L.J.; Dooyema, S.D.R.; Louwagie, E.J.; Baack, M.L. Maternal High-Fat Diet Impairs Cardiac Function in Offspring of Diabetic Pregnancy through Metabolic Stress and Mitochondrial Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H681–H692. [Google Scholar] [CrossRef] [Green Version]
- Ferey, J.L.A.; Boudoures, A.L.; Reid, M.; Drury, A.; Scheaffer, S.; Modi, Z.; Kovacs, A.; Pietka, T.; DeBosch, B.J.; Thompson, M.D.; et al. A Maternal High-Fat, High-Sucrose Diet Induces Transgenerational Cardiac Mitochondrial Dysfunction Independently of Maternal Mitochondrial Inheritance. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H1202–H1210. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, C.; Sun, M.; Maimaiti, R.; Ford, S.P.; Nathanielsz, P.W.; Ren, J.; Guo, W. Maternal Obesity Impairs Fetal Cardiomyocyte Contractile Function in Sheep. FASEB J. 2019, 33, 2587–2598. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, O.R.; Rosario, F.J.; Chan, J.; Cox, L.A.; Ferchaud-Roucher, V.; Zemski-Berry, K.A.; Reusch, J.E.B.; Keller, A.C.; Powell, T.L.; Jansson, T. Maternal Obesity Causes Fetal Cardiac Hypertrophy and Alters Adult Offspring Myocardial Metabolism in Mice. J. Physiol. 2022, 600, 3169–3191. [Google Scholar] [CrossRef]
- Khan, I.Y.; Taylor, P.D.; Dekou, V.; Seed, P.T.; Lakasing, L.; Graham, D.; Dominiczak, A.F.; Hanson, M.A.; Poston, L. Gender-Linked Hypertension in Offspring of Lard-Fed Pregnant Rats. Hypertension 2003, 41, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Eitmann, S.; Mátrai, P.; Németh, D.; Hegyi, P.; Lukács, A.; Bérczi, B.; Czumbel, L.M.; Kiss, I.; Gyöngyi, Z.; Varga, G.; et al. Maternal Overnutrition Elevates Offspring’s Blood Pressure—A Systematic Review and Meta-analysis. Paediatr. Perinat. Epidemiol. 2022, 36, 276–287. [Google Scholar] [CrossRef]
- Aceti, A.; Santhakumaran, S.; Logan, K.M.; Philipps, L.H.; Prior, E.; Gale, C.; Hyde, M.J.; Modi, N. The Diabetic Pregnancy and Offspring Blood Pressure in Childhood: A Systematic Review and Meta-Analysis. Diabetologia 2012, 55, 3114–3127. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal Obesity, Diabetes during Pregnancy and Epigenetic Mechanisms That Influence the Developmental Origins of Cardiometabolic Disease in the Offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef]
- Tobi, E.W.; Heijmans, B.T.; Kremer, D.; Putter, H.; Delemarre-van de Waal, H.A.; Finken, M.J.J.; Wit, J.M.; Slagboom, P.E. DNA Methylation of IGF2, GNASAS, INSIGF and LEP and Being Born Small for Gestational Age. Epigenetics 2011, 6, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA Methylation Differences after Exposure to Prenatal Famine Are Common and Timing- and Sex-Specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [Green Version]
- De Jong, K.A.; Barrand, S.; Wood-Bradley, R.J.; de Almeida, D.L.; Czeczor, J.K.; Lopaschuk, G.D.; Armitage, J.A.; McGee, S.L. Maternal High Fat Diet Induces Early Cardiac Hypertrophy and Alters Cardiac Metabolism in Sprague Dawley Rat Offspring. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 600–609. [Google Scholar] [CrossRef]
- Siddeek, B.; Mauduit, C.; Chehade, H.; Blin, G.; Liand, M.; Chindamo, M.; Benahmed, M.; Simeoni, U. Long-Term Impact of Maternal High-Fat Diet on Offspring Cardiac Health: Role of Micro-RNA Biogenesis. Cell Death Discov. 2019, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Shufelt, C.L.; Pacheco, C.; Tweet, M.S.; Miller, V.M. Sex-Specific Physiology and Cardiovascular Disease. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1065, pp. 433–454. [Google Scholar]
- Boonpattrawong, N.P.; Golbidi, S.; Tai, D.C.; Aleliunas, R.E.; Bernatchez, P.; Miller, J.W.; Laher, I.; Devlin, A.M. Exercise during Pregnancy Mitigates the Adverse Effects of Maternal Obesity on Adult Male Offspring Vascular Function and Alters One-Carbon Metabolism. Physiol. Rep. 2020, 8, e14582. [Google Scholar] [CrossRef]
- Schoonejans, J.M.; Ozanne, S.E. Developmental Programming by Maternal Obesity: Lessons from Animal Models. Diabet. Med. 2021, 38, e14694. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Chen, H.Z.; Wang, L.; Liu, D.P.; Hill, J.A.; Liu, Z.P. The Histone Trimethyllysine Demethylase JMJD2A Promotes Cardiac Hypertrophy in Response to Hypertrophic Stimuli in Mice. J. Clin. Investig. 2011, 121, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- TL, L.; CR, T.; CL, Y.; YP, W.; SH, K. Estrogen Receptor-β in Mitochondria: Implications for Mitochondrial Bioenergetics and Tumorigenesis. Ann. N. Y. Acad. Sci. 2015, 1350, 52–60. [Google Scholar] [CrossRef]
- Pauline, C.; Talbot, J.; Dolinsky, V.W.; Dolinsky, V.W. Sex Differences in the Developmental Origins of Cardiometabolic Disease Following Exposure to Maternal Obesity and Gestational Diabetes. Appl. Physiol. Nutr. Metab. 2019, 44, 687–695. [Google Scholar]
- Shvedova, M.; Anfinogenova, Y.; Popov, S.V.; Atochin, D.N. Connexins and Nitric Oxide inside and Outside Mitochondria: Significance for Cardiac Protection and Adaptation. Front. Physiol. 2018, 9, 479. [Google Scholar] [CrossRef] [Green Version]
- Kirca, M.; Kleinbongard, P.; Soetkamp, D.; Heger, J.; Csonka, C.; Ferdinandy, P.; Schulz, R. Interaction between Connexin 43 and Nitric Oxide Synthase in Mice Heart Mitochondria. J. Cell. Mol. Med. 2015, 19, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial Bioenergetics and Cardiolipin Alterations in Myocardial Ischemia/Reperfusion Injury: Implications for Pharmacological Cardioprotection. Am. J. Physiol. Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Smith, K.; Yu, Q.; Miller, C.; Singh, K.; Sen, C.K. Mitochondrial Connexin 43 in Sex-Dependent Myocardial Responses and Estrogen-Mediated Cardiac Protection Following Acute Ischemia/Reperfusion Injury. Basic Res Cardiol 2020, 115, 1. [Google Scholar] [CrossRef]
- Aroor, A.R.; Mandavia, C.H.; Sowers, J.R. Insulin Resistance and Heart Failure. Heart Fail. Clin. 2012, 8, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Turdi, S.; Ge, W.; Hu, N.; Bradley, K.M.; Wang, X.; Ren, J. Interaction between Maternal and Postnatal High Fat Diet Leads to a Greater Risk of Myocardial Dysfunction in Offspring via Enhanced Lipotoxicity, IRS-1 Serine Phosphorylation and Mitochondrial Defects. J. Mol. Cell. Cardiol. 2013, 55, 117–129. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, Q.; Halderson, S.J.; Arriola Apelo, S.I.; Jones, A.K.; Pillai, S.M.; Hoffman, M.L.; Reed, S.; Govoni, K.E.; Zinn, S.A.; et al. Maternal Overnutrition During Gestation in Sheep Alters Autophagy Associated Pathways in Offspring Heart. Front. Genet. 2022, 12, 742704. [Google Scholar] [CrossRef]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid Metabolism and Signaling in Cardiac Lipotoxicity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1513–1524. [Google Scholar] [CrossRef]
- Pantaleão, L.C.; Inzani, I.; Furse, S.; Loche, E.; Hufnagel, A.; Ashmore, T.; Blackmore, H.L.; Jenkins, B.; Carpenter, A.A.M.; Wilczynska, A.; et al. Maternal Diet-Induced Obesity during Pregnancy Alters Lipid Supply to Mouse E18.5 Fetuses and Changes the Cardiac Tissue Lipidome in a Sex-Dependent Manner. eLife 2022, 11, e69078. [Google Scholar] [CrossRef]
- Baker, P.R.; Patinkin, Z.; Shapiro, A.L.B.; De La Houssaye, B.A.; Woontner, M.; Boyle, K.E.; Vanderlinden, L.; Dabelea, D.; Friedman, J.E. Maternal Obesity and Increased Neonatal Adiposity Correspond with Altered Infant Mesenchymal Stem Cell Metabolism. JCI Insight 2017, 2, e94200. [Google Scholar] [CrossRef] [Green Version]
- Gyllenhammer, L.E.; Entringer, S.; Buss, C.; Wadhwa, P.D. Developmental Programming of Mitochondrial Biology: A Conceptual Framework and Review. Proc. Biol. Sci. 2020, 287, 20192713. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Burwell, L.S.; Brookes, P.S. Cardioprotection and Mitochondrial S-Nitrosation: Effects of S-Nitroso-2-Mercaptopropionyl Glycine (SNO-MPG) in Cardiac Ischemia-Reperfusion Injury. J. Mol. Cell. Cardiol. 2007, 42, 812–825. [Google Scholar] [CrossRef] [Green Version]
- La Morgia, C.; Maresca, A.; Caporali, L.; Valentino, M.L.; Carelli, V. Mitochondrial Diseases in Adults. J. Intern. Med. 2020, 287, 592–608. [Google Scholar] [CrossRef]
- Xue, Q.; Chen, F.; Zhang, H.; Liu, Y.; Chen, P.; Patterson, A.J.; Luo, J. Maternal High-Fat Diet Alters Angiotensin II Receptors and Causes Changes in Fetal and Neonatal Rats†. Biol. Reprod. 2019, 100, 1193–1203. [Google Scholar] [CrossRef]
- Bugger, H.; Schwarzer, M.; Chen, D.; Schrepper, A.; Amorim, P.A.; Schoepe, M.; Nguyen, T.D.; Mohr, F.W.; Khalimonchuk, O.; Weimer, B.C.; et al. Proteomic Remodelling of Mitochondrial Oxidative Pathways in Pressure Overload-Induced Heart Failure. Cardiovasc. Res. 2010, 85, 376–384. [Google Scholar] [CrossRef]
- Chiñas Merlin, A.; Gonzalez, K.; Mockler, S.; Perez, Y.; Jia, U.-T.A.; Chicco, A.J.; Ullevig, S.L.; Chung, E. Switching to a Standard Chow Diet at Weaning Improves the Effects of Maternal and Postnatal High-Fat and High-Sucrose Diet on Cardiometabolic Health in Adult Male Mouse Offspring. Metabolites 2022, 12, 563. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.D.; Sabey, K.H.; Knutson, A.J.; Gandy, T.C.T.; Louwagie, E.J.; Lauterboeck, L.; Mdaki, K.S.; Baack, M.L. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. Int. J. Mol. Sci. 2019, 20, 3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Han, X.; Fang, Z.; Che, L.; Nelson, J.; Yan, T.; Wu, D. Beneficial Effects of Dietary Fibre Supplementation of a High-Fat Diet on Fetal Development in Rats. Br. J. Nutr. 2011, 106, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired Glucose Homeostasis and Mitochondrial Abnormalities in Offspring of Rats Fed a Fat-Rich Diet in Pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, 134–139. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, L.; Tan, Y.; Zheng, Y.; Gui, Y. N-Acetylcysteine Protects Neonatal Mice from Ventricular Hypertrophy Induced by Maternal Obesity in a Sex-Specific Manner. Biomed. Pharmacother. 2021, 133, 110989. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide Activates Mitochondrial Uncoupling Proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Bhagavata Srinivasan, S.P.; Raipuria, M.; Bahari, H.; Kaakoush, N.O. Impacts of Diet and Exercise on Maternal Gut Microbiota Are Transferred to Offspring. Front. Endocrinol. 2018, 9, 716. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.D.; Gu, H.; Saunders, H.; Fiori, F.; Dalrymple, K.V.; Sethupathi, P.; Yamanouchi, L.; Miller, F.; Jones, B.; Vieira, M.C.; et al. Lifestyle Intervention in Obese Pregnancy and Cardiac Remodelling in 3-Year Olds: Children of the UPBEAT RCT. Int. J. Obes. 2022, 46, 2145–2155. [Google Scholar] [CrossRef]
- Ramalingam, L.; Menikdiwela, K.R.; Clevenger, S.; Eboh, T.; Allen, L.; Koboziev, I.; Scoggin, S.; Rashid, A.M.; Moussa, H.; Moustaid-Moussa, N. Maternal and Postnatal Supplementation of Fish Oil Improves Metabolic Health of Mouse Male Offspring. Obesity 2018, 26, 1740–1748. [Google Scholar] [CrossRef] [Green Version]
- Albert, B.B.; Vickers, M.H.; Gray, C.; Reynolds, C.M.; Segovia, S.A.; Derraik, J.G.B.; Garg, M.L.; Cameron-Smith, D.; Hofman, P.L.; Cutfield, W.S. Fish Oil Supplementation to Rats Fed High-Fat Diet during Pregnancy Prevents Development of Impaired Insulin Sensitivity in Male Adult Offspring. Sci. Rep. 2017, 7, 5595. [Google Scholar] [CrossRef]
- Satokar, V.V.; Vickers, M.H.; Reynolds, C.M.; Ponnampalam, A.P.; Firth, E.C.; Garg, M.L.; Barrett, C.J.; Cutfield, W.S.; Albert, B.B. Fish Oil Supplementation of Rats Fed a High Fat Diet during Pregnancy Improves Offspring Insulin Sensitivity. Front. Nutr. 2022, 9, 968443. [Google Scholar] [CrossRef]
- Gray, C.; Vickers, M.H.; Segovia, S.A.; Zhang, X.D.; Reynolds, C.M. A Maternal High Fat Diet Programmes Endothelial Function and Cardiovascular Status in Adult Male Offspring Independent of Body Weight, Which Is Reversed by Maternal Conjugated Linoleic Acid (CLA) Supplementation. PLoS ONE 2015, 10, e0115994. [Google Scholar] [CrossRef]
- Satokar, V.V.; Derraik, J.G.B.; Harwood, M.; Okesene-Gafa, K.; Beck, K.; Cameron-Smith, D.; Garg, M.L.; O’Sullivan, J.M.; Sundborn, G.; Pundir, S.; et al. Fish Oil Supplementation during Pregnancy and Postpartum in Mothers with Overweight and Obesity to Improve Body Composition and Metabolic Health during Infancy: A Double-Blind Randomized Controlled Trial. Am. J. Clin. Nutr. 2023, 117, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Rubini, E.; Schenkelaars, N.; Rousian, M.; Sinclair, K.D.; Wekema, L.; Faas, M.M.; Steegers-Theunissen, R.P.M.; Schoenmakers, S. Maternal Obesity during Pregnancy Leads to Derangements in One-Carbon Metabolism and the Gut Microbiota: Implications for Fetal Development and Offspring Wellbeing. Am. J. Obstet. Gynecol. 2022, 227, 392–400. [Google Scholar] [CrossRef]
- Maragkoudaki, X.; Naylor, M.; Papacleovoulou, G.; Stolarczyk, E.; Rees, D.; Pombo, J.M.; Abu-Hayyeh, S.; Czajka, A.; Howard, J.K.; Malik, A.N.; et al. Supplementation with a Prebiotic (Polydextrose) in Obese Mouse Pregnancy Improves Maternal Glucose Homeostasis and Protects against Offspring Obesity. Int. J. Obes. 2020, 44, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Naomi, R.; Rusli, R.N.M.; Othman, F.; Balan, S.S.; Abidin, A.Z.; Embong, H.; Teoh, S.H.; Jasni, A.S.; Jumidil, S.H.; Matraf, K.S.Y.B.; et al. Elateriospermum Tapos Yogurt Supplement in Maternal Obese Dams during Pregnancy Modulates the Body Composition of F1 Generation. Nutrients 2023, 15, 1258. [Google Scholar] [CrossRef]
- Santos, A.C.C.; Amaro, L.B.R.; Jorge, A.H.B.; Lelis, S.D.F.; Lelis, D.D.F.; Guimarães, A.L.S.; Santos, S.H.S.; Andrade, J.M.O. Curcumin Improves Metabolic Response and Increases Expression of Thermogenesis-Associated Markers in Adipose Tissue of Male Offspring from Obese Dams. Mol. Cell. Endocrinol. 2023, 563, 111840. [Google Scholar] [CrossRef]
- Lentjes, M.A.H. The Balance between Food and Dietary Supplements in the General Population. Proc. Nutr. Soc. 2019, 78, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Wooltorton, E. Too Much of a Good Thing? Toxic Effects of Vitamin and Mineral Supplements. CMAJ 2003, 169, 47–48. [Google Scholar]
- Pinckard, K.; Baskin, K.K.; Stanford, K.I. Effects of Exercise to Improve Cardiovascular Health. Front. Cardiovasc. Med. 2019, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burelle, Y.; Wambolt, R.B.; Grist, M.; Parsons, H.L.; Chow, J.C.F.; Antler, C.; Bonen, A.; Keller, A.; Dunaway, G.A.; Popov, K.M.; et al. Regular Exercise Is Associated with a Protective Metabolic Phenotype in the Rat Heart. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, 1055–1063. [Google Scholar] [CrossRef]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise Training Boosts ENOS-Dependent Mitochondrial Biogenesis in Mouse Heart: Role in Adaptation of Glucose Metabolism. Am. J. Physiol. Endocrinol. Metab. 2014, 306, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riehle, C.; Wende, A.R.; Zhu, Y.; Oliveira, K.J.; Pereira, R.O.; Jaishy, B.P.; Bevins, J.; Valdez, S.; Noh, J.; Kim, B.J.; et al. Insulin Receptor Substrates Are Essential for the Bioenergetic and Hypertrophic Response of the Heart to Exercise Training. Mol. Cell. Biol. 2014, 34, 3450–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, S.R.; Hawley, J.A. Update on the Effects of Physical Activity on Insulin Sensitivity in Humans. BMJ Open Sport Exerc. Med. 2017, 2, e000143. [Google Scholar] [CrossRef] [Green Version]
- Johan, E.H.; Baer, L.A.; Stanford, K.I. Maternal Exercise Improves the Metabolic Health of Adult Offspring. Physiol. Behav. 2018, 29, 164–177. [Google Scholar] [CrossRef]
- Vega, C.C.; Reyes-Castro, L.A.; Bautista, C.J.; Larrea, F.; Nathanielsz, P.W.; Zambrano, E. Exercise in Obese Female Rats Has Beneficial Effects on Maternal and Male and Female Offspring Metabolism. Int. J. Obes. 2015, 39, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.D.; Nicole Blaize, A.; Fletcher, J.A.; Pearson, K.J.; Donkin, S.S.; Newcomer, S.C.; Rector, R.S. Gestational Exercise Protects Adult Male Offspring from High-Fat Diet-Induced Hepatic Steatosis. J. Hepatol. 2016, 64, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Torrens, C.; Ethirajan, P.; Bruce, K.D.; Cagampang, F.R.A.; Siow, R.C.M.; Hanson, M.A.; Byrne, C.D.; Mann, G.E.; Clough, G.F. Interaction between Maternal and Offspring Diet to Impair Vascular Function and Oxidative Balance in High Fat Fed Male Mice. PLoS ONE 2012, 7, e50671. [Google Scholar] [CrossRef] [Green Version]
- Grasemann, C.; Herrmann, R.; Starschinova, J.; Gertsen, M.; Palmert, M.R.; Grasemann, H. Effects of Fetal Exposure to High-Fat Diet or Maternal Hyperglycemia on L-Arginine and Nitric Oxide Metabolism in Lung. Nutr. Diabetes 2017, 7, e244. [Google Scholar] [CrossRef] [Green Version]
- Resende, A.C.; Emiliano, A.F.; Cordeiro, V.S.C.; de Bem, G.F.; de Cavalho, L.C.R.M.; de Oliveira, P.R.B.; Neto, M.L.; Costa, C.A.; Boaventura, G.T.; de Moura, R.S. Grape Skin Extract Protects against Programmed Changes in the Adult Rat Offspring Caused by Maternal High-Fat Diet during Lactation. J. Nutr. Biochem. 2013, 24, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Beeson, J.H.; Blackmore, H.L.; Carr, S.K.; Dearden, L.; Duque-Guimarães, D.E.; Kusinski, L.C.; Pantaleão, L.C.; Pinnock, A.G.; Aiken, C.E.; Giussani, D.A.; et al. Maternal Exercise Intervention in Obese Pregnancy Improves the Cardiovascular Health of the Adult Male Offspring. Mol. Metab. 2018, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Nyrnes, S.A.; Garnæs, K.K.; Salvesen, Ø.; Timilsina, A.S.; Moholdt, T.; Ingul, C.B. Cardiac Function in Newborns of Obese Women and the Effect of Exercise during Pregnancy. A Randomized Controlled Trial. PLoS ONE 2019, 13, e0197334. [Google Scholar] [CrossRef]
- Harris, J.E.; Pinckard, K.M.; Wright, K.R.; Baer, L.A.; Arts, P.J.; Abay, E.; Shettigar, V.K.; Lehnig, A.C.; Robertson, B.; Madaris, K.; et al. Exercise-Induced 3′Sialyllactose in Breastmilk Is A Critical Mediator to Improve Metabolic Health and Cardiac Function in Mice Offspring. Physiol. Behav. 2020, 176, 139–148. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diniz, M.S.; Grilo, L.F.; Tocantins, C.; Falcão-Pires, I.; Pereira, S.P. Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb. Metabolites 2023, 13, 845. https://doi.org/10.3390/metabo13070845
Diniz MS, Grilo LF, Tocantins C, Falcão-Pires I, Pereira SP. Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb. Metabolites. 2023; 13(7):845. https://doi.org/10.3390/metabo13070845
Chicago/Turabian StyleDiniz, Mariana S., Luís F. Grilo, Carolina Tocantins, Inês Falcão-Pires, and Susana P. Pereira. 2023. "Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb" Metabolites 13, no. 7: 845. https://doi.org/10.3390/metabo13070845
APA StyleDiniz, M. S., Grilo, L. F., Tocantins, C., Falcão-Pires, I., & Pereira, S. P. (2023). Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb. Metabolites, 13(7), 845. https://doi.org/10.3390/metabo13070845