
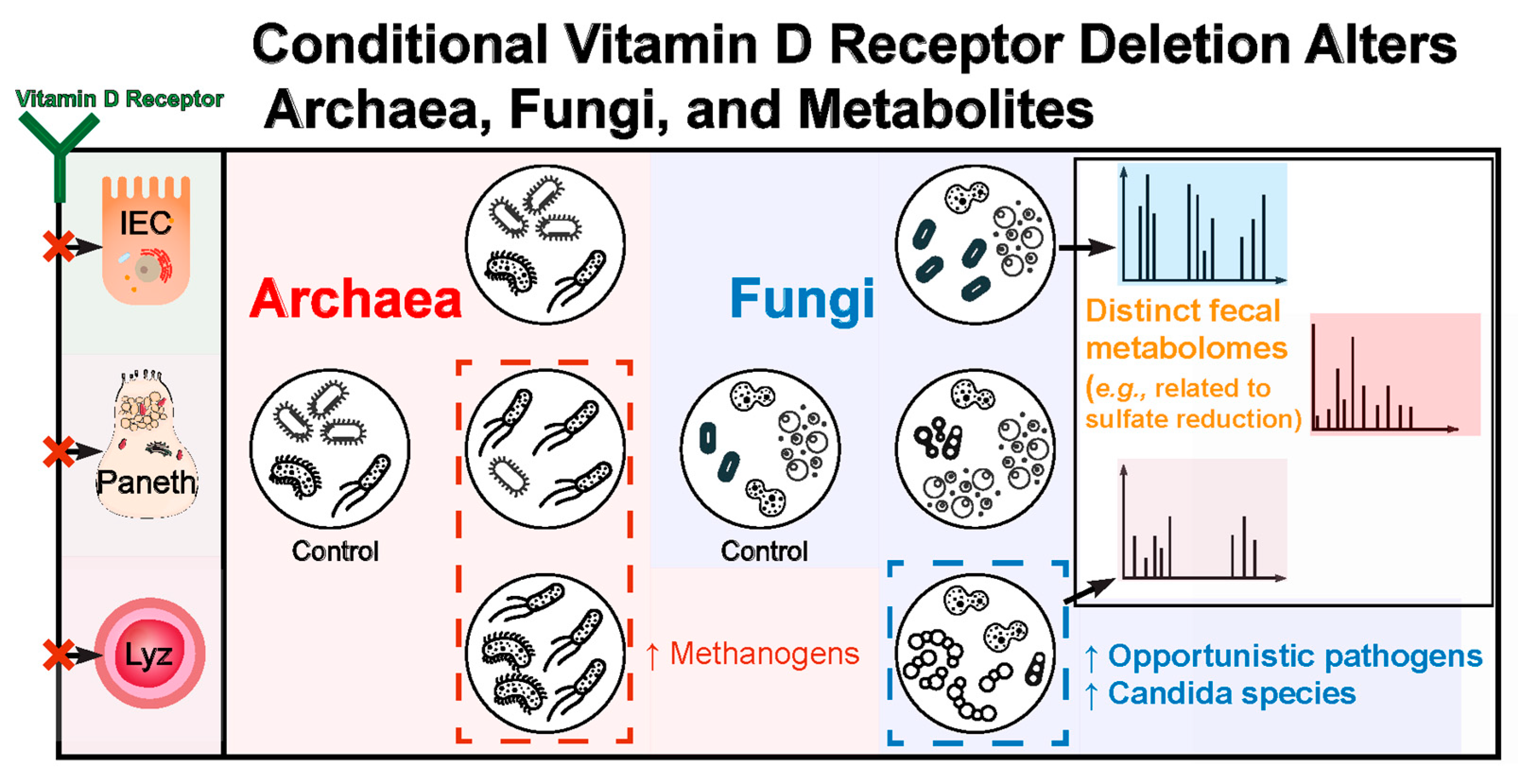
Conditional Vitamin D Receptor Deletion Induces Fungal and Archaeal Dysbiosis and Altered Metabolites


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Shotgun Sequencing
2.3. Metabolomics
2.4. Statistical Analysis
2.5. Microbiome Data Analysis
2.5.1. Alpha Diversity
2.5.2. Beta Diversity
2.5.3. Differential Abundance
2.5.4. Correlation Analysis
2.5.5. Fungi Annotation
2.5.6. Functional Analysis
2.5.7. Metabolomics Analysis
3. Results
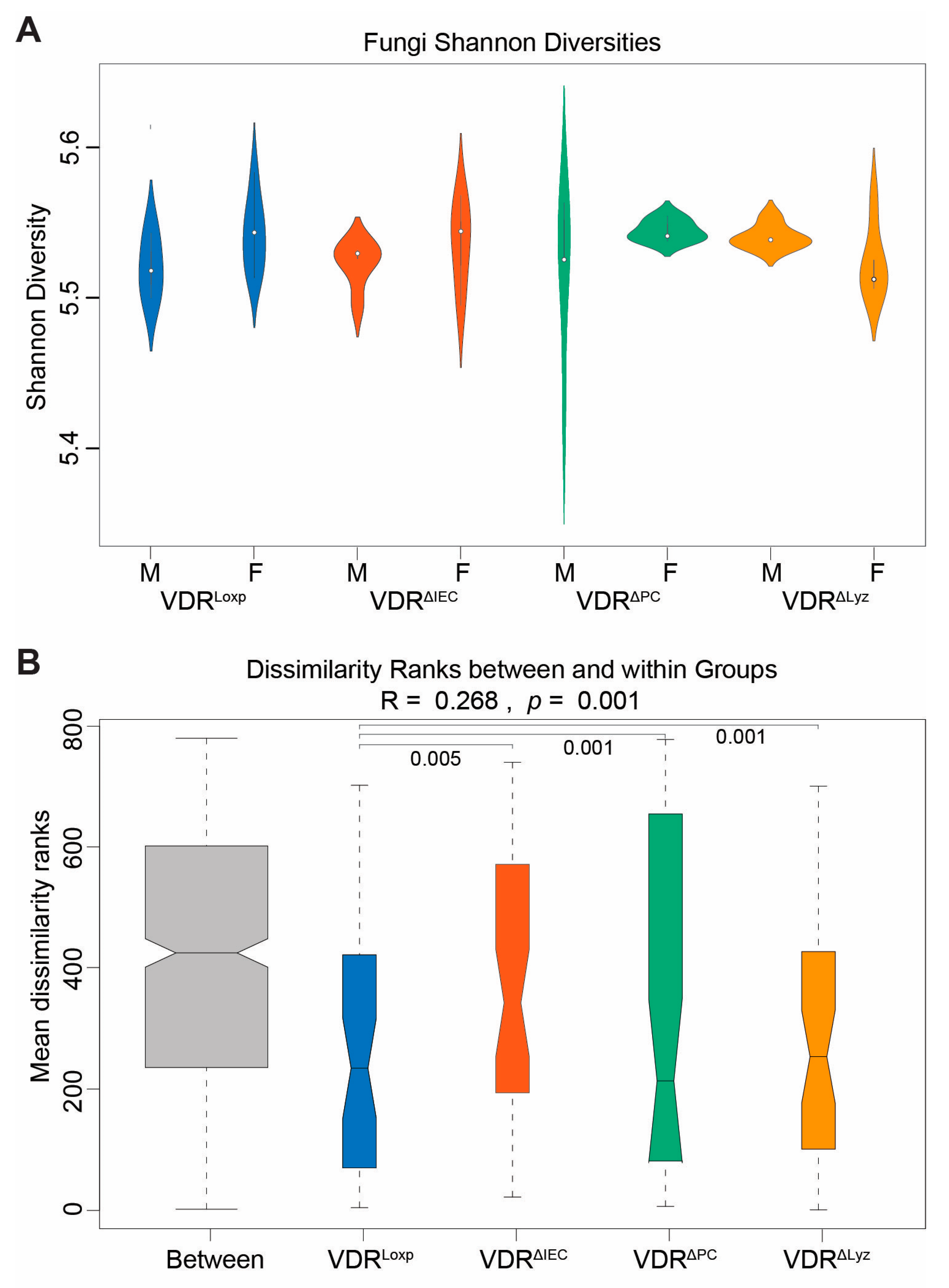
3.1. Conditional VDR KO Alters Fungal Beta Diversity
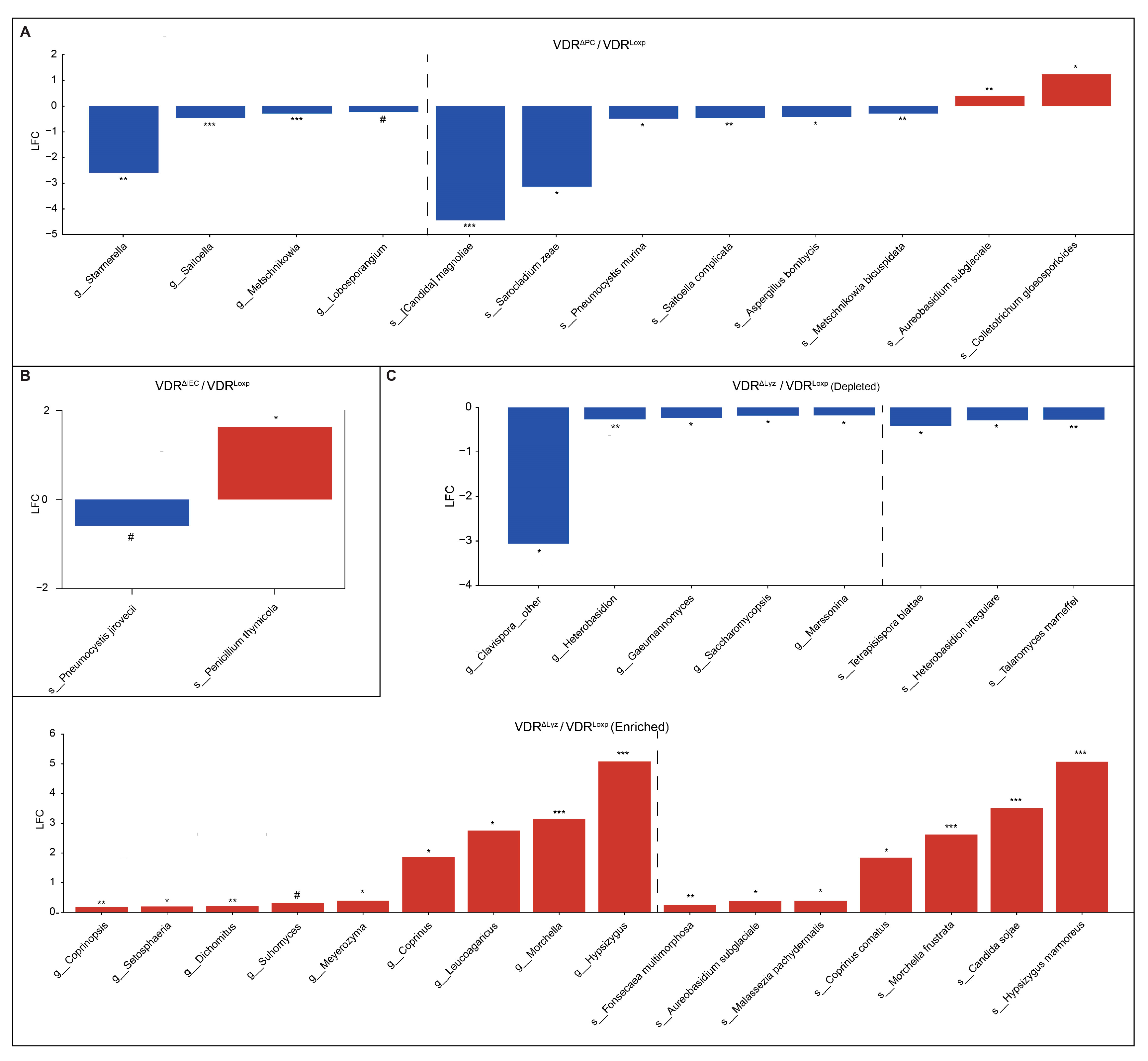
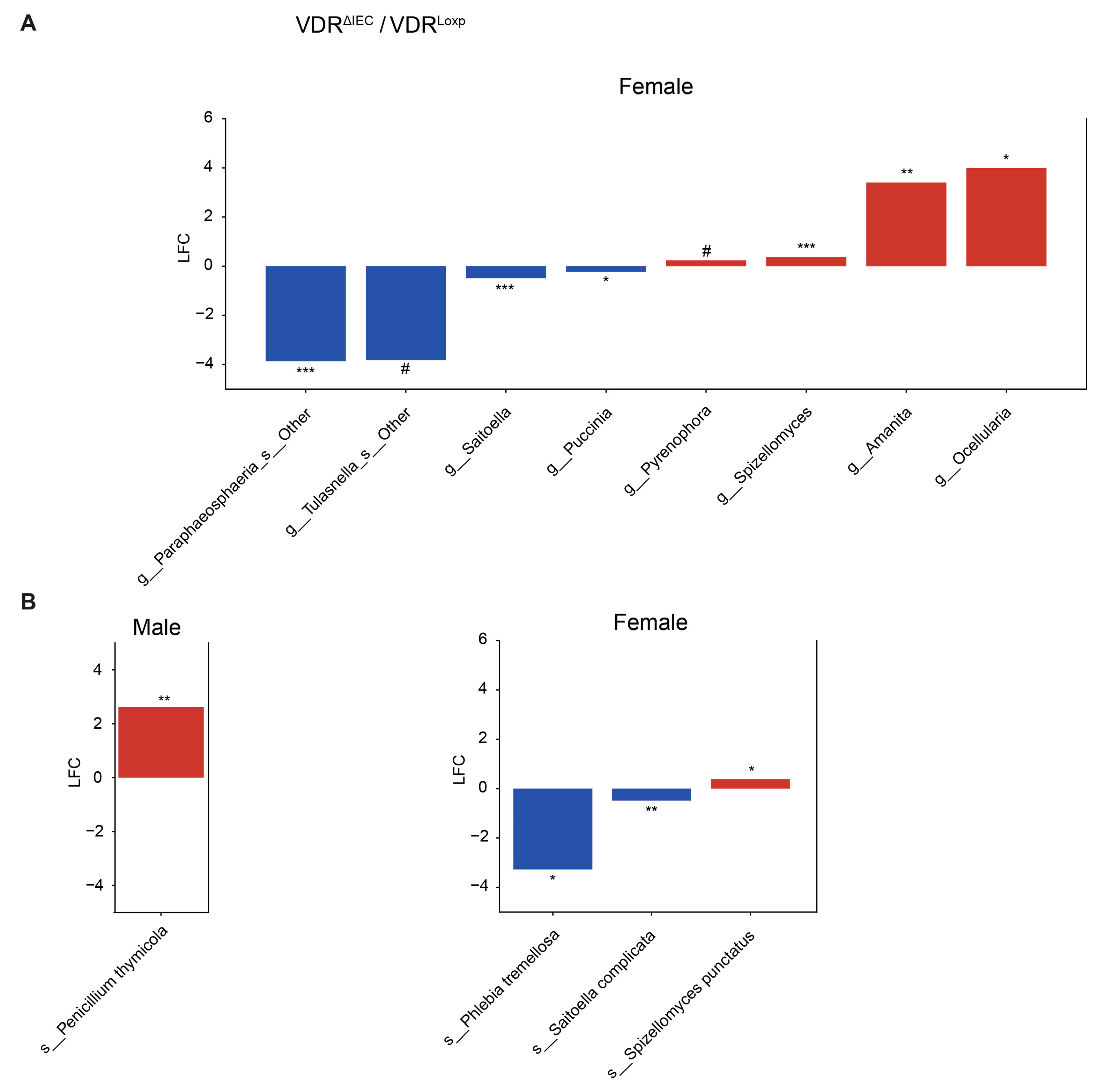
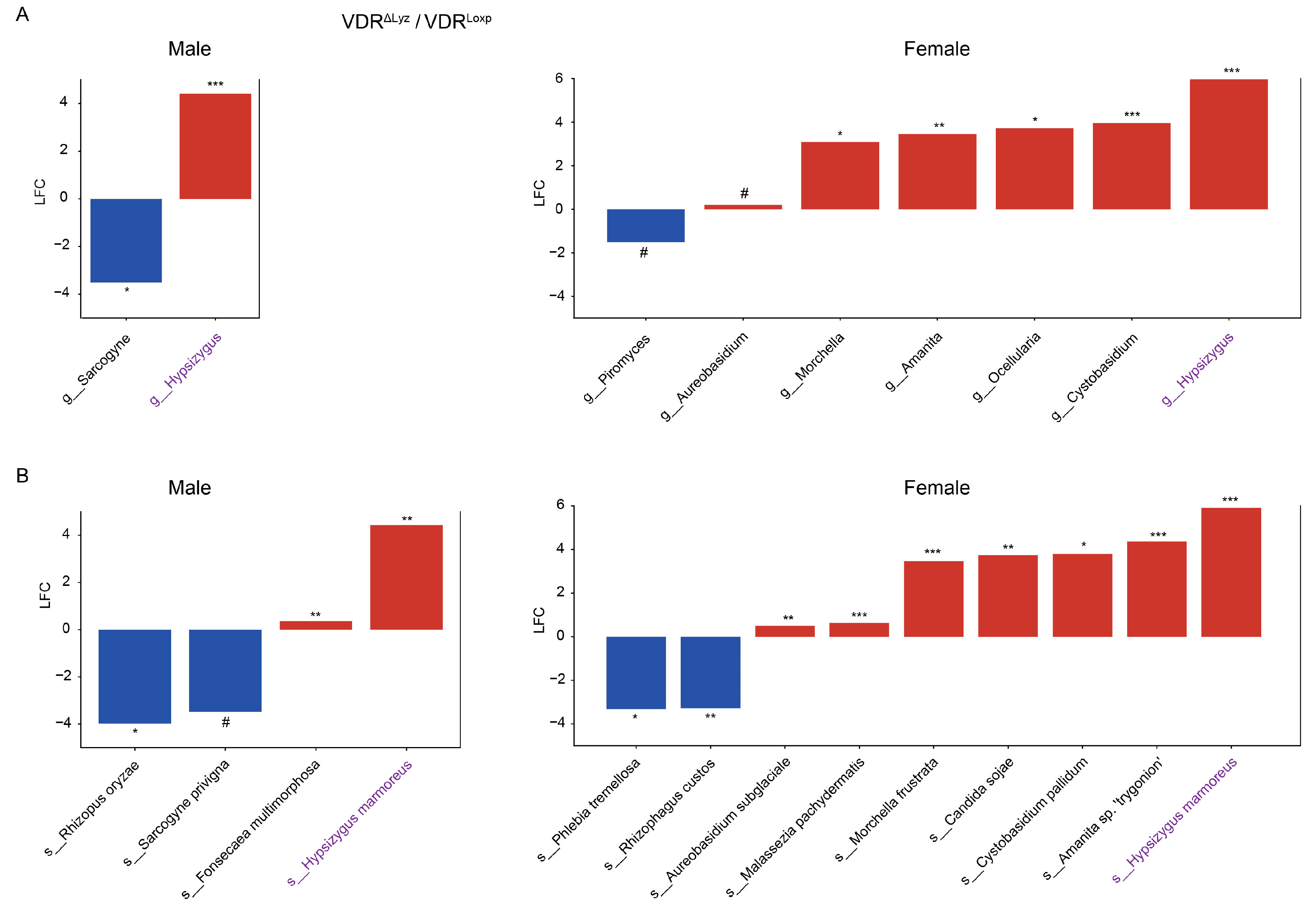
3.2. Differentially Abundant Fungi Vary by VDR KO in Different Tissues
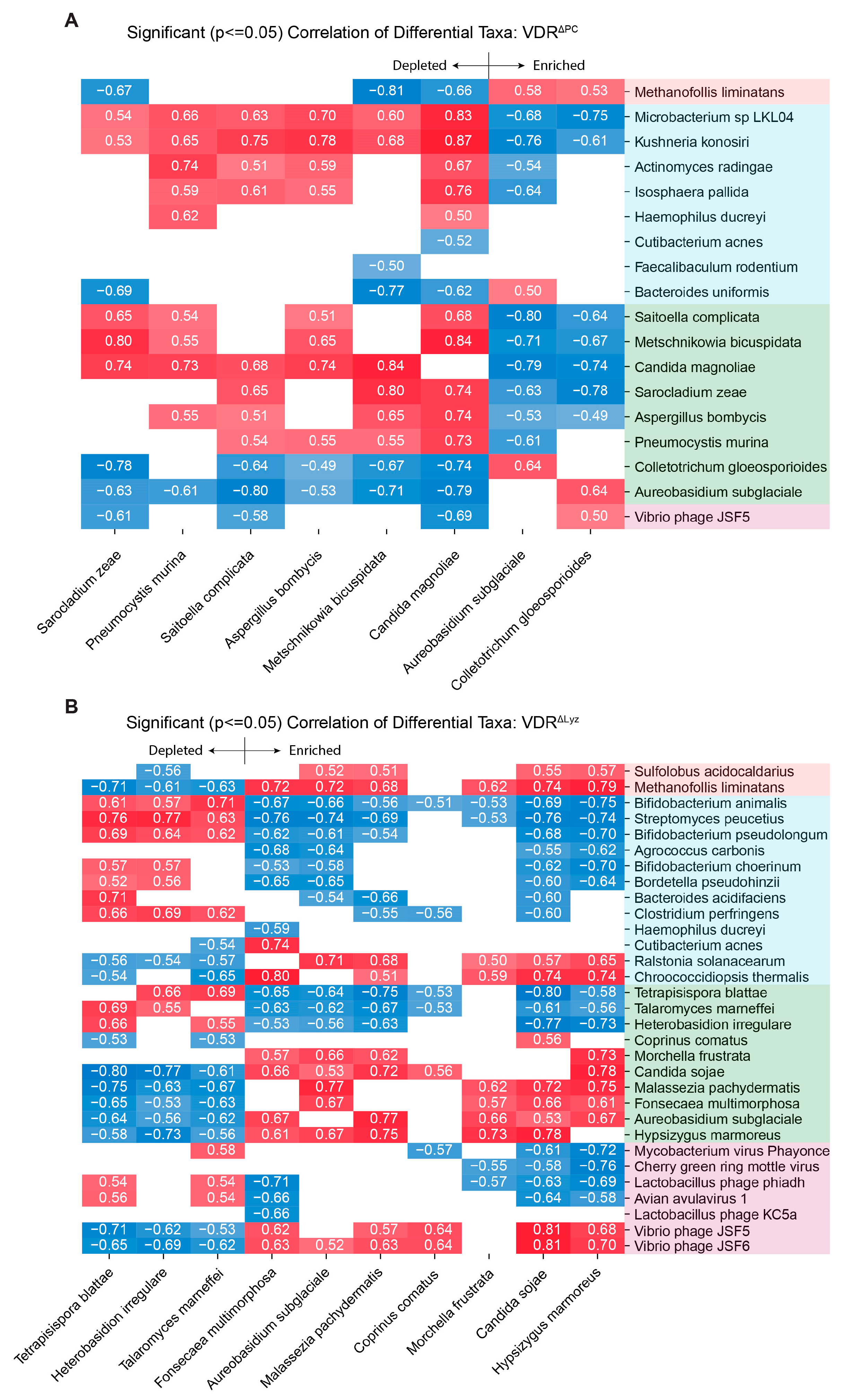
3.3. Significant Fungal Correlations Exist with Archaea, Bacteria, and Viruses
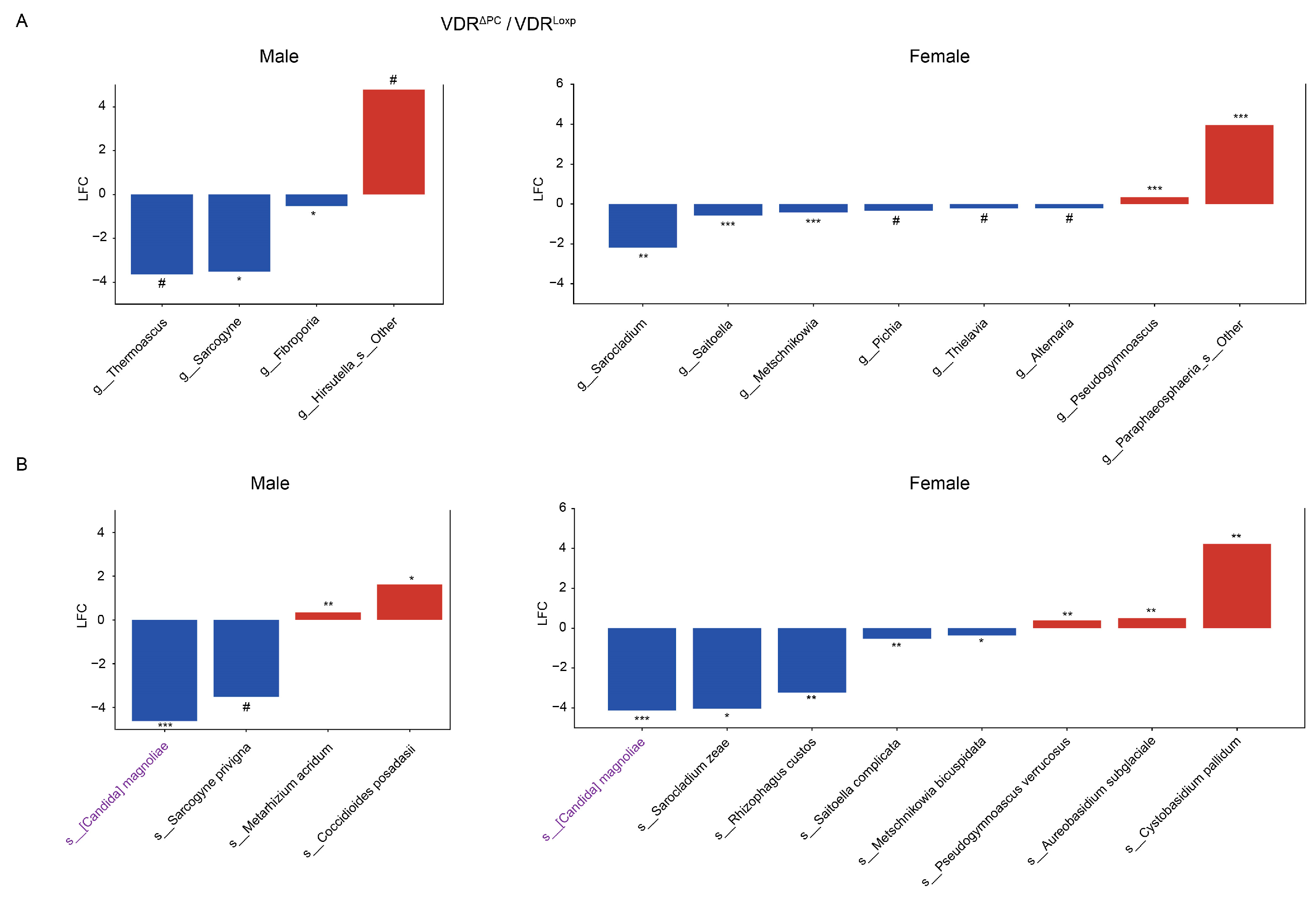
3.4. Sex Differences of Fungal Dysbiosis in the VDR KO Mice
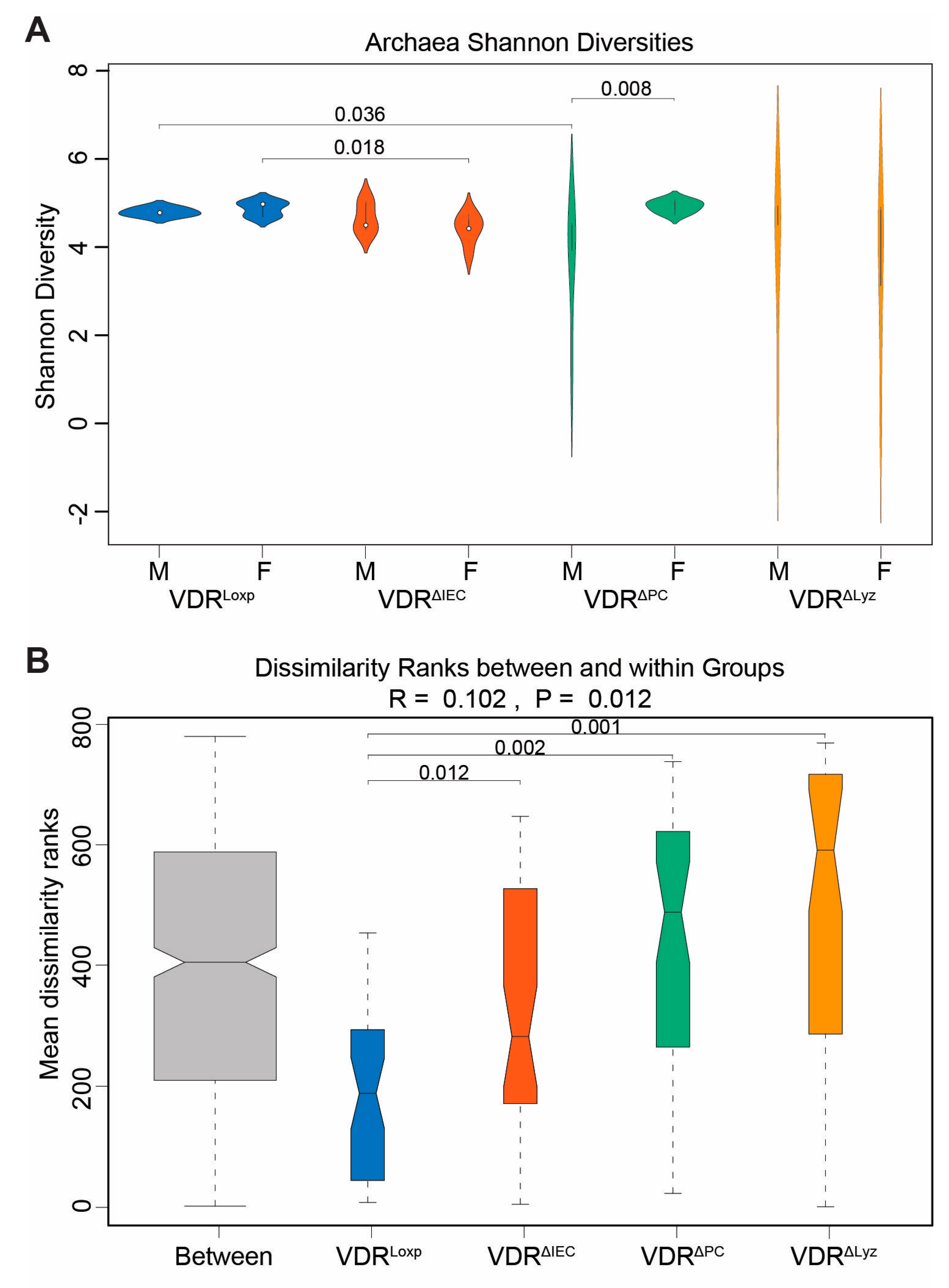
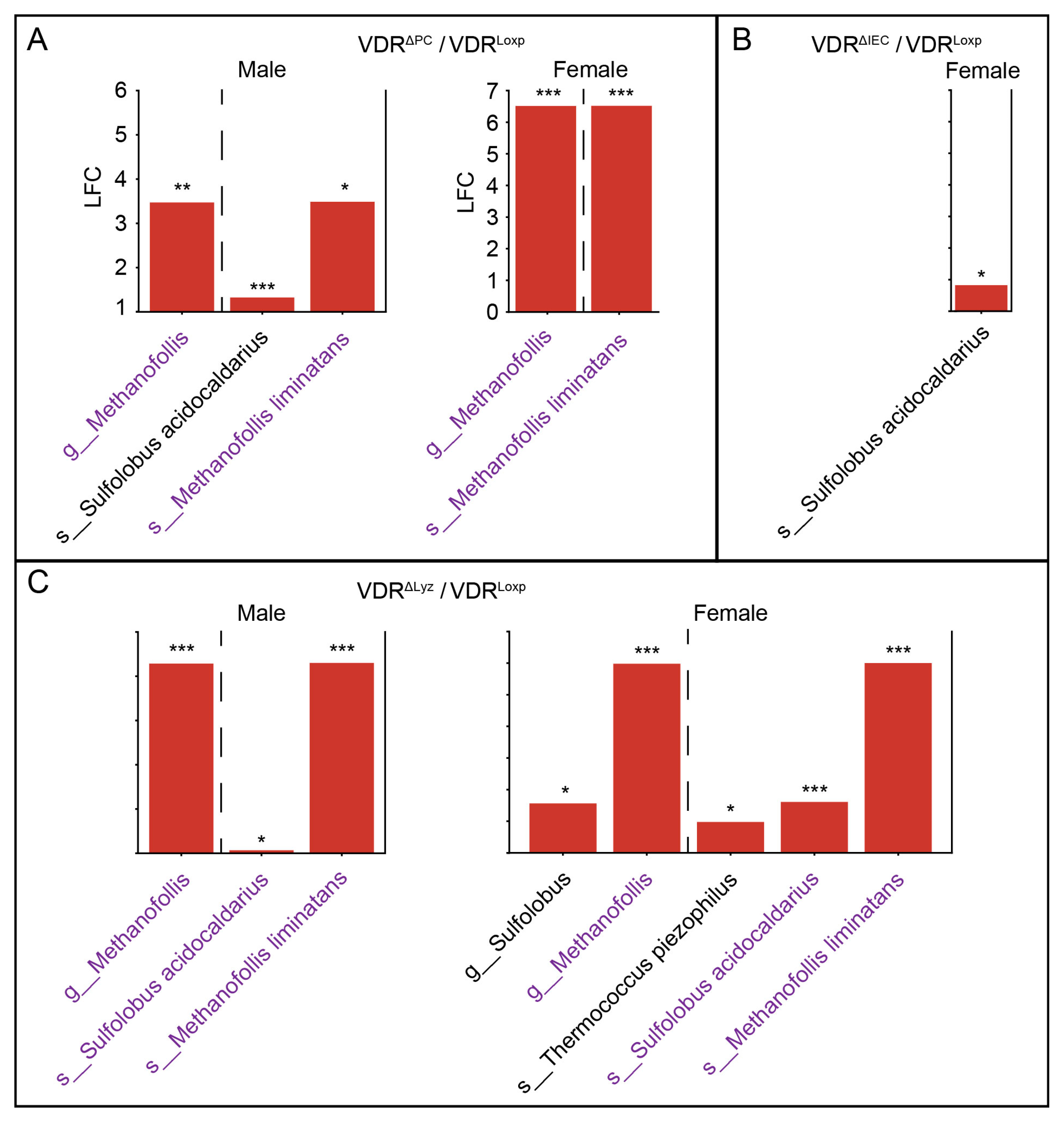
3.5. VDR Knockouts Alter Archaeal Alpha and Beta Diversities
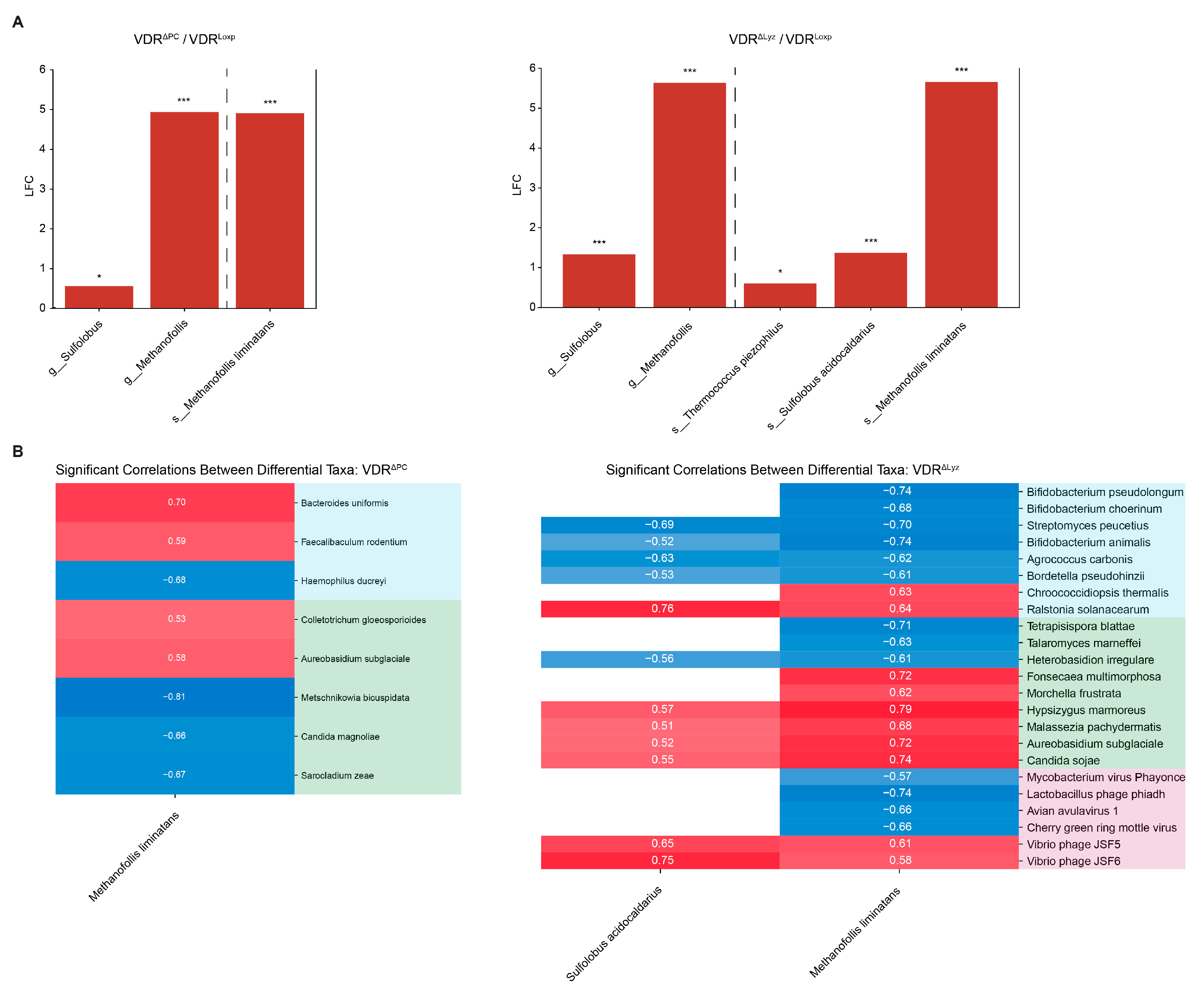
3.6. Common Set of Abundant Archaea in the VDRΔPC and VDRΔLyz Groups
3.7. Signficant Archaeal Correlations Exist with Bacteria, Fungi, and Viruses
3.8. The Archaeal Dysbiosis Caused by VDR Deletions Is Sex Dependent
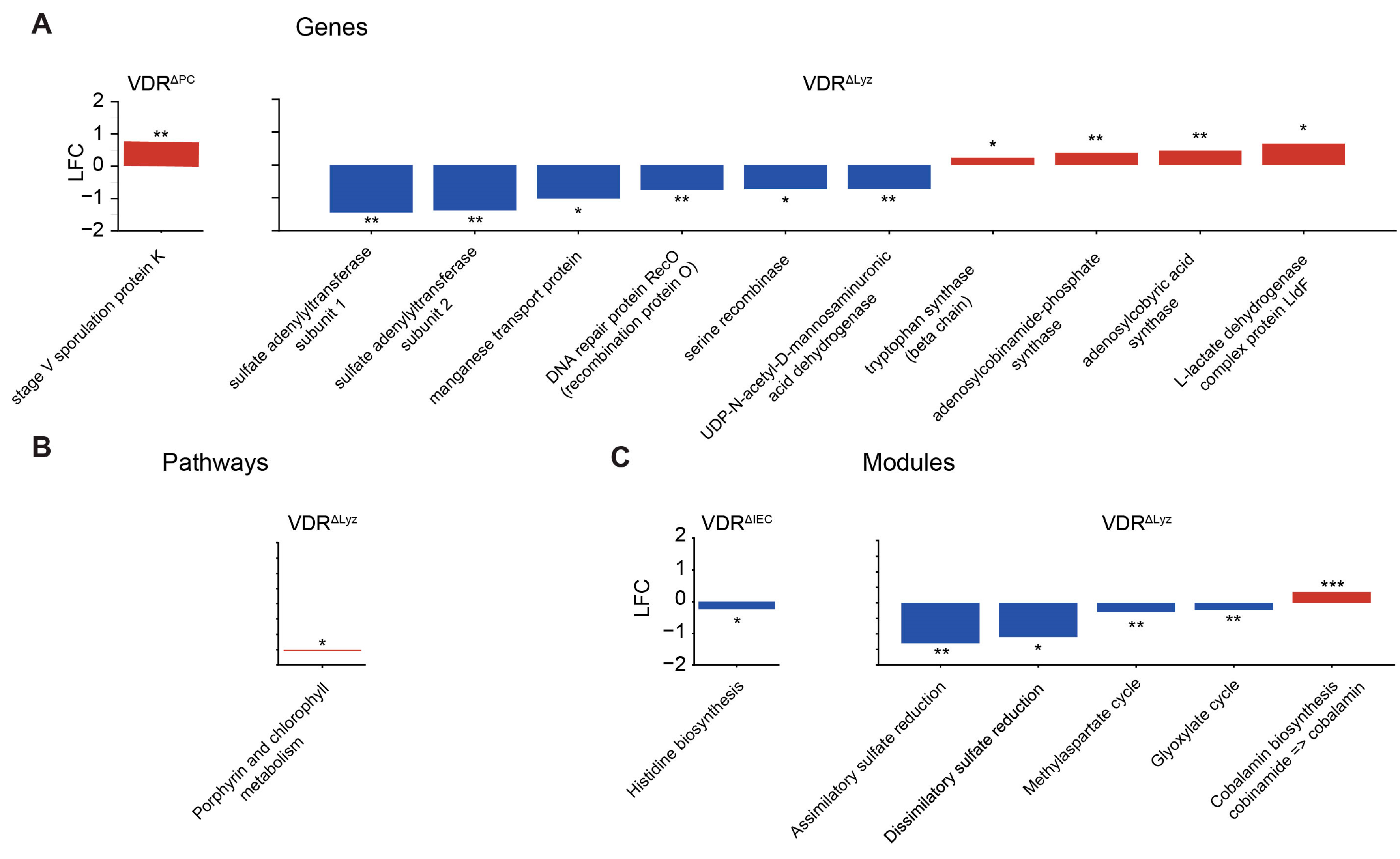
3.9. Functional Metagenomics
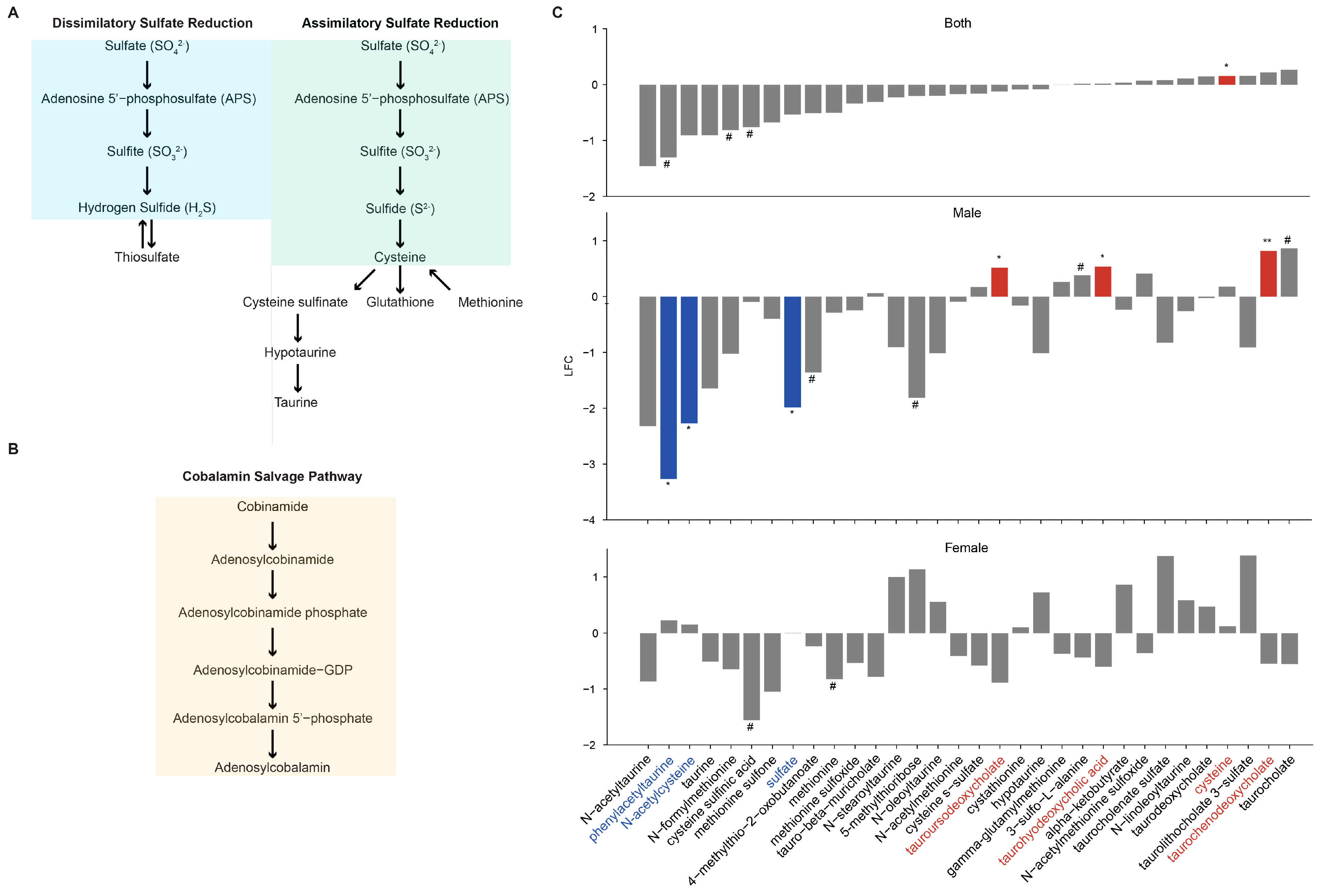
3.10. Metabolomic Signiture Related to VDR KO in Myeloid Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bakke, D.; Sun, J. Ancient Nuclear Receptor VDR with New Functions: Microbiome and Inflammation. Inflamm. Bowel Dis. 2018, 24, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Mishra, A.; Ashraf, M.Z. Emerging Role of Vitamin D and its Associated Molecules in Pathways Related to Pathogenesis of Thrombosis. Biomolecules 2019, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Barbáchano, A.; Fernández-Barral, A.; Ferrer-Mayorga, G.; Costales-Carrera, A.; Larriba, M.J.; Muñoz, A. The endocrine vitamin D system in the gut. Mol. Cell. Endocrinol. 2017, 453, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Chen, Y.; Shi, Y.; Liu, T.; Cao, Y.; Tang, Y.; Ge, X.; Nie, H.; Zheng, C.; Li, Y.C. 1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway. Inflamm. Bowel Dis. 2015, 21, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yu, Y.; Lu, J.; Lu, L.; Zhang, Y.G.; Xia, Y.; Sun, J.; Claud, E.C. The Impact of Maternal Probiotics on Intestinal Vitamin D Receptor Expression in Early Life. Biomolecules 2023, 13, 847. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.H.; Li, Y.; Rogers, C.J.; Cantorna, M.T. Vitamin D Regulates the Gut Microbiome and Protects Mice from Dextran Sodium Sulfate–Induced Colitis123. J. Nutr. 2013, 143, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Cannell, J.J.; Vieth, R.; Umhau, J.C.; Holick, M.F.; Grant, W.B.; Madronich, S.; Garland, C.F.; Giovannucci, E. Epidemic influenza and vitamin D. Epidemiol. Infect. 2006, 134, 1129–1140. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Xia, Y.; Sun, J. Imbalance of the intestinal virome and altered viral-bacterial interactions caused by a conditional deletion of the vitamin D receptor. Gut Microbes 2021, 13, 1957408. [Google Scholar] [CrossRef]
- Jin, D.; Wu, S.; Zhang, Y.-g.; Lu, R.; Xia, Y.; Dong, H.; Sun, J. Lack of Vitamin D Receptor Causes Dysbiosis and Changes the Functions of the Murine Intestinal Microbiome. Clin. Ther. 2015, 37, 996–1009. [Google Scholar] [CrossRef]
- Rashed, R.; Valcheva, R.; Dieleman, L.A. Manipulation of Gut Microbiota as a Key Target for Crohn’s Disease. Front. Med. 2022, 9, 887044. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Rakoff-Nahoum, S. Understanding Competition and Cooperation within the Mammalian Gut Microbiome. Curr. Biol. 2019, 29, R538–R544. [Google Scholar] [CrossRef] [PubMed]
- Kuziel, G.A.; Rakoff-Nahoum, S. The gut microbiome. Curr. Biol. 2022, 32, R257–R264. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.K.; Yong, V.C.; Chong, P.P.; Amin Nordin, S.; Basir, R.; Abdullah, M. Mycobiome in the Gut: A Multiperspective Review. Mediat. Inflamm. 2020, 2020, e9560684. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Aschenbrenner, D.; Yoo, J.Y.; Zuo, T. The gut mycobiome in health, disease, and clinical applications in association with the gut bacterial microbiome assembly. Lancet Microbe 2022, 3, e969–e983. [Google Scholar] [CrossRef] [PubMed]
- Seed, P.C. The Human Mycobiome. Cold Spring Harb. Perspect. Med. 2015, 5, a019810. [Google Scholar] [CrossRef] [PubMed]
- Tiew, P.Y.; Mac Aogain, M.; Ali, N.A.t.B.M.; Thng, K.X.; Goh, K.; Lau, K.J.X.; Chotirmall, S.H. The Mycobiome in Health and Disease: Emerging Concepts, Methodologies and Challenges. Mycopathologia 2020, 185, 207–231. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.O.; Claud, E.C.; Chen, D.; Chang, E.B.; Carmeliet, G.; et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2015, 64, 1082–1094. [Google Scholar] [CrossRef]
- Houshyar, Y.; Massimino, L.; Lamparelli, L.A.; Danese, S.; Ungaro, F. Going beyond Bacteria: Uncovering the Role of Archaeome and Mycobiome in Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 783295. [Google Scholar] [CrossRef]
- Matijašić, M.; Meštrović, T.; Čipčić Paljetak, H.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef]
- Lurie-Weinberger, M.N.; Gophna, U. Archaea in and on the Human Body: Health Implications and Future Directions. PLoS Pathog. 2015, 11, e1004833. [Google Scholar] [CrossRef] [PubMed]
- Hoegenauer, C.; Hammer, H.F.; Mahnert, A.; Moissl-Eichinger, C. Methanogenic archaea in the human gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.B.; Rostami, E.; Sephay, A.A.; Shahrokh, S.; Balaii, H.; Aghdaei, H.A.; Zali, M.R. Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb. Pathog. 2018, 117, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Wu, W.K.K.; Wong, S.H.; Sung, J.J.Y.; Yu, J. Altered Gut Archaea Composition and Interaction with Bacteria Are Associated with Colorectal Cancer. Gastroenterology 2020, 159, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Mathlouthi, N.E.H.; Oumarou Hama, H.; Belguith, I.; Charfi, S.; Boudawara, T.; Lagier, J.C.; Ammar Keskes, L.; Grine, G.; Gdoura, R. Colorectal Cancer Archaeome: A Metagenomic Exploration, Tunisia. Curr. Issues Mol. Biol. 2023, 45, 7572–7581. [Google Scholar] [CrossRef] [PubMed]
- Kos, C.H. Methods in Nutrition Science: Cre/loxP System for Generating Tissue-specific Knockout Mouse Models. Nutr. Rev. 2004, 62, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Sun, J.; Xia, Y.; Chen, D.-G. Statistical Analysis of Microbiome Data With R. ICSA Book Series in Statistics; Springer: Singapore, 2018; Volume 847. [Google Scholar]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Chatterjee, I.; Lu, R.; Zhang, Y.; Zhang, J.; Dai, Y.; Xia, Y.; Sun, J. Vitamin D receptor promotes healthy microbial metabolites and microbiome. Sci. Rep. 2020, 10, 7340. [Google Scholar] [CrossRef]
- Sherwin, W.B.; Jabot, F.; Rush, R.; Rossetto, M. Measurement of biological information with applications from genes to landscapes. Mol. Ecol. 2006, 15, 2857–2869. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J. Alpha Diversity. In Bioinformatic and Statistical Analysis of Microbiome Data: From Raw Sequences to Advanced Modeling with QIIME 2 and R; Springer International Publishing: Cham, Switzerland, 2023; pp. 289–333. [Google Scholar]
- Xia, Y.; Sun, J. (Eds.) Beta Diversity Metrics and Ordination. In Bioinformatic and Statistical Analysis of Microbiome Data: From Raw Sequences to Advanced Modeling with QIIME 2 and R; Springer International Publishing: Cham, Switzerland, 2023; pp. 335–395. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J. Statistical Testing of Beta Diversity. In Bioinformatic and Statistical Analysis of Microbiome Data: From Raw Sequences to Advanced Modeling with QIIME 2 and R; Springer International Publishing: Cham, Switzerland, 2023; pp. 397–433. [Google Scholar]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.A.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 2022, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, T. Package ‘misty’, 0.5.4; 2023. Available online: https://cran.r-project.org/web/packages/misty/index.html (accessed on 1 October 2023).
- Suhr, M.J.; Hallen-Adams, H.E. The human gut mycobiome: Pitfalls and potentials--a mycologists perspective. Mycologia 2015, 107, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Lavrinienko, A.; Scholier, T.; Bates, S.T.; Miller, A.N.; Watts, P.C. Defining gut mycobiota for wild animals: A need for caution in assigning authentic resident fungal taxa. Anim. Microbiome 2021, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Suhr, M.J.; Banjara, N.; Hallen-Adams, H.E. Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett. Appl. Microbiol. 2016, 62, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Iliev, I.D.; Brown, J.; Underhill, D.M.; Funari, V.A. Mycobiome: Approaches to analysis of intestinal fungi. J. Immunol. Methods 2015, 421, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Mantzoukas, S.; Kitsiou, F.; Natsiopoulos, D.; Eliopoulos, P.A. Entomopathogenic Fungi: Interactions and Applications. Encyclopedia 2022, 2, 646–656. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Kuo, M.; Dewsbury, D.R.; O’Donnell, K.; Carter, M.C.; Rehner, S.A.; Moore, J.D.; Moncalvo, J.M.; Canfield, S.A.; Stephenson, S.L.; Methven, A.S.; et al. Taxonomic revision of true morels (Morchella) in Canada and the United States. Mycologia 2012, 104, 1159–1177. [Google Scholar] [CrossRef]
- Kiran, M.; Khan, J.; Sher, H.; Pfister, D.H.; Khalid, A.N. Amanita griseofusca: A new species of Amanita in section Vaginatae from Malam Jabba, Swat, Pakistan. Phytotaxa 2018, 364, 181. [Google Scholar] [CrossRef]
- Zhang, J.j.; Shi, L.; Chen, H.; Sun, Y.q.; Zhao, M.w.; Ren, A.; Chen, M.j.; Wang, H.; Feng, Z.y. An efficient Agrobacterium-mediated transformation method for the edible mushroom Hypsizygus marmoreus. Microbiol. Res. 2014, 169, 741–748. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, J.; Tang, Q.; Liu, Y.; Zhang, A.; Pan, Y. Structural elucidation of a neutral fucogalactan from the mycelium of Coprinus comatus. Carbohydr. Res. 2006, 341, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Mahesh, G.; Priya, M.; Singh, R.U.S.; Manjunatha, L. Chapter 43-Hirsutella. In Beneficial Microbes in Agro-Ecology; Amaresan, N., Senthil Kumar, M., Annapurna, K., Kumar, K., Sankaranarayanan, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 817–831. [Google Scholar]
- Wolfe, K.H.; Armisén, D.; Proux-Wera, E.; ÓhÉigeartaigh, S.S.; Azam, H.; Gordon, J.L.; Byrne, K.P. Clade- and species-specific features of genome evolution in the Saccharomycetaceae. FEMS Yeast Res. 2015, 15, fov035. [Google Scholar] [CrossRef] [PubMed]
- Kamga, S.F.; Ndjomatchoua, F.T.; Guimapi, R.A.; Klingen, I.; Tchawoua, C.; Hjelkrem, A.-G.R.; Thunes, K.H.; Kakmeni, F.M. The effect of climate variability in the efficacy of the entomopathogenic fungus Metarhizium acridum against the desert locust Schistocerca gregaria. Sci. Rep. 2022, 12, 7535. [Google Scholar] [CrossRef] [PubMed]
- Sieverding, E.; Da Silva, G.A.; Berndt, R.; Oehl, F. Rhizoglomus, a new genus of the Glomeraceae. Mycotaxon 2015, 129, 373–386. [Google Scholar] [CrossRef]
- Kemp, N.D.; Vaughan, M.M.; McCormick, S.P.; Brown, J.A.; Bakker, M.G. Sarocladium zeae is a systemic endophyte of wheat and an effective biocontrol agent against Fusarium head blight. Biol. Control 2020, 149, 104329. [Google Scholar] [CrossRef]
- De Graeve, M.; De Maeseneire, S.L.; Roelants, S.L.K.W.; Soetaert, W. Starmerella bombicola, an industrially relevant, yet fundamentally underexplored yeast. FEMS Yeast Res. 2018, 18, foy072. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-H.; Lee, D.-H.; Oh, Y.-J.; Han, K.-C.; Ryu, Y.-W.; Seo, J.-H. Selective utilization of fructose to glucose by Candida magnoliae, an erythritol producer. Appl. Biochem. Biotechnol. 2006, 131, 870–879. [Google Scholar] [CrossRef]
- van der Merwe, M.; Ericson, L.; Walker, J.; Thrall, P.H.; Burdon, J.J. Evolutionary relationships among species of Puccinia and Uromyces (Pucciniaceae, Uredinales) inferred from partial protein coding gene phylogenies. Mycol. Res. 2007, 111, 163–175. [Google Scholar] [CrossRef]
- Gonthier, P.; Anselmi, N.; Capretti, P.; Bussotti, F.; Feducci, M.; Giordano, L.; Honorati, T.; Lione, G.; Luchi, N.; Michelozzi, M.; et al. An integrated approach to control the introduced forest pathogen Heterobasidion irregulare in Europe. For. Int. J. For. Res. 2014, 87, 471–481. [Google Scholar] [CrossRef]
- Gautam, A.K. Colletotrichum gloeosporioides: Biology, Pathogenicity and Management in India. J. Plant Physiol. Pathol. 2014, 2–11. [Google Scholar] [CrossRef]
- Badalyan, S.M.; Szafranski, K.; Hoegger, P.J.; Navarro-González, M.; Majcherczyk, A.; Kües, U. New Armenian Wood-Associated Coprinoid Mushrooms: Coprinopsis strossmayeri and Coprinellus aff. radians. Diversity 2011, 3, 136–154. [Google Scholar] [CrossRef]
- Kuuskeri, J.; Häkkinen, M.; Laine, P.; Smolander, O.-P.; Tamene, F.; Miettinen, S.; Nousiainen, P.; Kemell, M.; Auvinen, P.; Lundell, T. Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: Growth on spruce wood and decay effect on lignocellulose. Biotechnol. Biofuels 2016, 9, 192. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.; Kocourková, J. Sarcogyne praetermissa (Acarosporaceae), a New Calcicolous Lichen Species from Europe, with a Key to the European Sarcogyne Species. Heia 2018, 31, 133–139. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Cejnar, J.; Treml, J.; Dordevic, D.; Kollar, P.; Vitezova, M. Recent Advances in Metabolic Pathways of Sulfate Reduction in Intestinal Bacteria. Cells 2020, 9, 698. [Google Scholar] [CrossRef]
- Teigen, L.M.; Geng, Z.; Sadowsky, M.J.; Vaughn, B.P.; Hamilton, M.J.; Khoruts, A. Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis. Nutrients 2019, 11, 931. [Google Scholar] [CrossRef]
- Fang, H.; Kang, J.; Zhang, D. Microbial production of vitamin B(12): A review and future perspectives. Microb. Cell Fact. 2017, 16, 15. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Tang, C.; He, Q.; Li, N.; Li, J. Dysbiosis of Gut Fungal Microbiota is Associated with Mucosal Inflammation in Crohn’s Disease. J. Clin. Gastroenterol. 2014, 48, 513–523. [Google Scholar] [CrossRef]
- Zhai, C.-D.; Zheng, J.-J.; An, B.-C.; Huang, H.-F.; Tan, Z.-C. Intestinal microbiota composition in patients with amyotrophic lateral sclerosis: Establishment of bacterial and archaeal communities analyses. Chin. Med. J. 2019, 132, 1815–1822. [Google Scholar] [CrossRef]
- Limon, J.J.; Skalski, J.H.; Underhill, D.M. Commensal Fungi in Health and Disease. Cell Host Microbe 2017, 22, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Leonardi, I. Fungal dysbiosis: Immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 2017, 17, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Bakke, D.; Chatterjee, I.; Agrawal, A.; Dai, Y.; Sun, J. Regulation of Microbiota by Vitamin D Receptor: A Nuclear Weapon in Metabolic Diseases. Nucl. Recept. Res. 2018, 5, 101377. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, C.A. Inflammation and gastrointestinal Candida colonization. Curr. Opin. Microbiol. 2011, 14, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Nkamga, V.D.; Henrissat, B.; Drancourt, M. Archaea: Essential inhabitants of the human digestive microbiota. Hum. Microbiome J. 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Shivvers, D.W.; Brock, T.D. Oxidation of Elemental Sulfur by Sulfolobus acidocaldarius. J. Bacteriol. 1973, 114, 706–710. [Google Scholar] [CrossRef]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef]
- Walker, A.; Schmitt-Kopplin, P. The role of fecal sulfur metabolome in inflammatory bowel diseases. Int. J. Med. Microbiol. 2021, 311, 151513. [Google Scholar] [CrossRef]
- Candela, M.; Perna, F.; Carnevali, P.; Vitali, B.; Ciati, R.; Gionchetti, P.; Rizzello, F.; Campieri, M.; Brigidi, P. Interaction of probiotic Lactobacillus and Bifidobacterium strains with human intestinal epithelial cells: Adhesion properties, competition against enteropathogens and modulation of IL-8 production. Int. J. Food Microbiol. 2008, 125, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Bakke, D.S.; Zhang, J.; Zhang, Y.; Ogbu, D.; Xia, Y.; Sun, J. Myeloid vitamin D receptor regulates Paneth cells and microbial homeostasis. FASEB J. 2023, 37, e22957. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhang, Y.G.; Xia, Y.; Zhang, J.; Kaser, A.; Blumberg, R.; Sun, J. Paneth Cell Alertness to Pathogens Maintained by Vitamin D Receptors. Gastroenterology 2021, 160, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.K.; Paul, A.; Jahan, R.; Jannat, K.; Bondhon, T.A.; Hasan, A.; Nissapatorn, V.; Pereira, M.L.; Wilairatana, P.; Rahmatullah, M. Probiotics and Amelioration of Rheumatoid Arthritis: Significant Roles of Lactobacillus casei and Lactobacillus acidophilus. Microorganisms 2021, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- Naser, I.B.; Hoque, M.M.; Nahid, M.A.; Tareq, T.M.; Rocky, M.K.; Faruque, S.M. Analysis of the CRISPR-Cas system in bacteriophages active on epidemic strains of Vibrio cholerae in Bangladesh. Sci. Rep. 2017, 7, 14880. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Manser, C.; Pittet, V.; Vavricka, S.R.; Biedermann, L.; on behalf of Swiss IBDnet, an official working group of the Swiss Society of Gastroenterology. Gender Differences in Inflammatory Bowel Disease. Digestion 2020, 101, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Strati, F.; Di Paola, M.; Stefanini, I.; Albanese, D.; Rizzetto, L.; Lionetti, P.; Calabrò, A.; Jousson, O.; Donati, C.; Cavalieri, D.; et al. Age and Gender Affect the Composition of Fungal Population of the Human Gastrointestinal Tract. Front. Microbiol. 2016, 7, 1227. [Google Scholar] [CrossRef]
- Li, Q.; Chan, H.; Liu, W.X.; Liu, C.A.; Zhou, Y.; Huang, D.; Wang, X.; Li, X.; Xie, C.; Liu, W.Y.; et al. Carnobacterium maltaromaticum boosts intestinal vitamin D production to suppress colorectal cancer in female mice. Cancer Cell 2023, 41, 1450–1465. [Google Scholar] [CrossRef]
- Maruvada, P.; Leone, V.; Kaplan, L.M.; Chang, E.B. The Human Microbiome and Obesity: Moving beyond Associations. Cell Host Microbe 2017, 22, 589–599. [Google Scholar] [CrossRef]
- Levitt, M.D.; Furne, J.K.; Kuskowski, M.; Ruddy, J. Stability of Human Methanogenic Flora over 35 Years and a Review of Insights Obtained from Breath Methane Measurements. Clin. Gastroenterol. Hepatol. 2006, 4, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sulfate Adenylyltransferase Subunit 1 | Sulfate Adenylyltransferase Subunit 2 | Adenosylcobyric Acid Synthase | Adenosylcobinamide-Phosphate Synthase | |
|---|---|---|---|---|
| K00956 | K00957 | K02232 | K02227 | |
| Thermococcus piezophilus | - | - | + | + |
| Bordetella pseudohinzii | + | + | - | - |
| Chroococcidiopsis thermalis | - | - | + | + |
| Sulfurovum lithotrophicum | + | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claypool, D.J.; Zhang, Y.-G.; Xia, Y.; Sun, J. Conditional Vitamin D Receptor Deletion Induces Fungal and Archaeal Dysbiosis and Altered Metabolites. Metabolites 2024, 14, 32. https://doi.org/10.3390/metabo14010032
Claypool DJ, Zhang Y-G, Xia Y, Sun J. Conditional Vitamin D Receptor Deletion Induces Fungal and Archaeal Dysbiosis and Altered Metabolites. Metabolites. 2024; 14(1):32. https://doi.org/10.3390/metabo14010032
Chicago/Turabian StyleClaypool, Duncan J., Yong-Guo Zhang, Yinglin Xia, and Jun Sun. 2024. "Conditional Vitamin D Receptor Deletion Induces Fungal and Archaeal Dysbiosis and Altered Metabolites" Metabolites 14, no. 1: 32. https://doi.org/10.3390/metabo14010032
APA StyleClaypool, D. J., Zhang, Y. -G., Xia, Y., & Sun, J. (2024). Conditional Vitamin D Receptor Deletion Induces Fungal and Archaeal Dysbiosis and Altered Metabolites. Metabolites, 14(1), 32. https://doi.org/10.3390/metabo14010032