

Celastrol Stabilizes Glycolipid Metabolism in Hepatic Steatosis by Binding and Regulating the Peroxisome Proliferator-Activated Receptor γ Signaling Pathway

Abstract
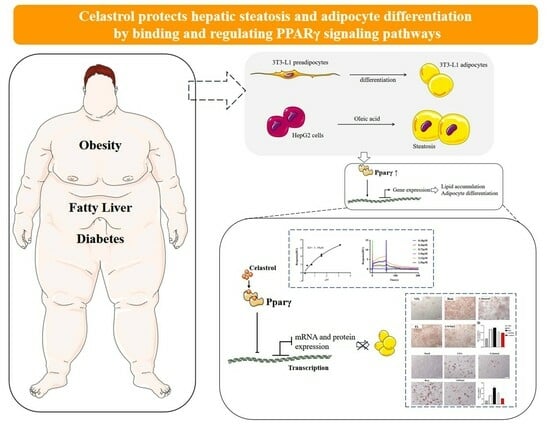
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Culture and Steatosis of HepG2 Cells
2.3. Adipocytic Differentiation of 3T3-L1 Cells
2.4. Cell Viability Assay
2.5. Oil Red O Staining
2.6. PPARγ Agonist Reverse Experiment
2.7. Glucose Absorption Assay
2.8. Glucose Consumption Assay
2.9. Triglyceride Content Assay
2.10. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.11. Ligand Binding Assay
2.12. Statistical Analysis
3. Results
3.1. Celastrol Protects against Oleic-Acid-Induced Fatty Liver
3.2. Celastrol Directly Binds PPARγ and Inhibits Its Transcriptional Activity
3.3. Rosiglitazone Can Reverse the Fatty Liver Protective Effects of Celastrol on Fatty Liver Tissue
3.4. Celastrol Does Not Influence Glucose Uptake Function
3.5. Celastrol Inhibits Adipocyte Differentiation of 3T3-L1 Adipocytes
3.6. Celastrol Alters the Gene Expression Profile Associated with Adipocyte Differentiation in 3T3-L1 Adipocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Li, Y.; Luo, S.; Ye, J.; Lu, Z.; Li, X.; Lu, H. The HIF-2α/PPARα pathway is essential for liraglutide-alleviated, lipid-induced hepatic steatosis. Biomed. Pharmacother. 2021, 140, 111778. [Google Scholar] [CrossRef]
- Cioffi, F.; Giacco, A.; Petito, G.; de Matteis, R.; Senese, R.; Lombardi, A.; de Lange, P.; Moreno, M.; Goglia, F.; Lanni, A.; et al. Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding. Genes 2022, 13, 315. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.R.; Graffeo, C.S.; Rehman, A.; Fallon, N.C.; Zambirinis, C.P.; Ochi, A.; Barilla, R.; Jamal, M.; Deutsch, M.; Greco, S.; et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013, 58, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Park, S.; Jang, S.; Cho, H.H.; Lee, S.; You, S.; Kim, S.H.; Moon, H.S. Cascade regulation of PPARγ2 and C/EBPα signaling pathways by celastrol impairs adipocyte differentiation and stimulates lipolysis in 3T3-L1 adipocytes. Metab. Clin. Exp. 2016, 65, 646–654. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Sirén, J.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef]
- Choi, Y.; Song, M.J.; Jung, W.J.; Jeong, H.; Park, S.; Yang, B.; Lee, E.C.; Joo, J.S.; Choi, D.; Koo, S.H.; et al. Liver-Specific Deletion of Mouse CTCF Leads to Hepatic Steatosis via Augmented PPARγ Signaling. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1761–1787. [Google Scholar] [CrossRef]
- Baumann, A.; Burger, K.; Brandt, A.; Staltner, R.; Jung, F.; Rajcic, D.; Lorenzo Pisarello, M.J.; Bergheim, I. GW9662, a peroxisome proliferator-activated receptor gamma antagonist, attenuates the development of non-alcoholic fatty liver disease. Metab. Clin. Exp. 2022, 133, 155233. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; He, B.; Yu, M.; Shi, M.; Zhu, Y.; Zhang, Z.; Bo, P. Treatment with celastrol protects against obesity through suppression of galanin-induced fat intake and activation of PGC-1α/GLUT4 axis-mediated glucose consumption. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Zhao, J.; Zhao, W.; Zhang, X.; Song, Q.; Shen, Y.; Shum, H.C.; Wang, Y.; Rong, J. Celastrol-loaded lactosylated albumin nanoparticles attenuate hepatic steatosis in non-alcoholic fatty liver disease. J. Control. Release Off. J. Control. Release Soc. 2022, 347, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, H.J.; Yu, J.; Li, T.; Han, Y. Protective effect of celastrol on type 2 diabetes mellitus with nonalcoholic fatty liver disease in mice. Food Sci. Nutr. 2020, 8, 6207–6216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Geng, C.; Liu, X.; Li, M.; Gao, M.; Liu, X.; Fang, F.; Chang, Y. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Mol. Metab. 2017, 6, 138–147. [Google Scholar] [CrossRef]
- Ramadori, G.; Fujikawa, T.; Fukuda, M.; Anderson, J.; Morgan, D.A.; Mostoslavsky, R.; Stuart, R.C.; Perello, M.; Vianna, C.R.; Nillni, E.A.; et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab. 2010, 12, 78–87. [Google Scholar] [CrossRef]
- Semple, R.K.; Chatterjee, V.K.; O’Rahilly, S. PPAR gamma and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef]
- Kahn, B.B.; McGraw, T.E. Rosiglitazone, PPARγ, and type 2 diabetes. N. Engl. J. Med. 2010, 363, 2667–2669. [Google Scholar] [CrossRef]
- McGuire, D.K.; Inzucchi, S.E. New drugs for the treatment of diabetes mellitus: Part I: Thiazolidinediones and their evolving cardiovascular implications. Circulation 2008, 117, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.; McGee, S.L. 3T3-L1 adipocytes display phenotypic characteristics of multiple adipocyte lineages. Adipocyte 2015, 4, 295–302. [Google Scholar] [CrossRef]
- Paternostro, R.; Trauner, M. Current treatment of non-alcoholic fatty liver disease. J. Intern. Med. 2022, 292, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Martin, A.; Zhang, X.; Farowski, F.; Wisplinghoff, H.; Vehreschild, M.J.G.T.; Krawczyk, M.; Nowag, A.; Kretzschmar, A.; Scholz, C.; et al. Combined analysis of gut microbiota, diet and PNPLA3 polymorphism in biopsy-proven non-alcoholic fatty liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2021, 41, 1576–1591. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Zappavigna, S.; Vanacore, D.; Lama, S.; Potenza, N.; Russo, A.; Ferranti, P.; Dallio, M.; Federico, A.; Loguercio, C.; Sperlongano, P.; et al. Silybin-Induced Apoptosis Occurs in Parallel to the Increase of Ceramides Synthesis and miRNAs Secretion in Human Hepatocarcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2190. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Z.; Wang, S.; Guo, H.; Dong, S.; Wang, T.; Zhang, L.; Jiang, Z. Emodin ameliorates hepatic steatosis through endoplasmic reticulum-stress sterol regulatory element-binding protein 1c pathway in liquid fructose-feeding rats. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2016, 46, E105–E117. [Google Scholar] [CrossRef]
- Zhu, X.; Bian, H.; Gao, X. The Potential Mechanisms of Berberine in the Treatment of Nonalcoholic Fatty Liver Disease. Molecules 2016, 21, 1336. [Google Scholar] [CrossRef]
- Pisonero-Vaquero, S.; Martínez-Ferreras, Á.; García-Mediavilla, M.V.; Martínez-Flórez, S.; Fernández, A.; Benet, M.; Olcoz, J.L.; Jover, R.; González-Gallego, J.; Sánchez-Campos, S. Quercetin ameliorates dysregulation of lipid metabolism genes via the PI3K/AKT pathway in a diet-induced mouse model of nonalcoholic fatty liver disease. Mol. Nutr. Food Res. 2015, 59, 879–893. [Google Scholar] [CrossRef]
- Kessoku, T.; Imajo, K.; Honda, Y.; Kato, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. Resveratrol ameliorates fibrosis and inflammation in a mouse model of nonalcoholic steatohepatitis. Sci. Rep. 2016, 6, 22251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Ge, H.Y.; Gu, Y.J.; Cao, F.F.; Yang, C.X.; Uzan, G.; Peng, B.; Zhang, D.H. Celastrol reverses palmitic acid (PA)-caused TLR4-MD2 activation-dependent insulin resistance via disrupting MD2-related cellular binding to PA. J. Cell. Physiol. 2018, 233, 6814–6824. [Google Scholar] [CrossRef]
- Zhou, H.; Ma, C.; Wang, C.; Gong, L.; Zhang, Y.; Li, Y. Research progress in use of traditional Chinese medicine monomer for treatment of non-alcoholic fatty liver disease. Eur. J. Pharmacol. 2021, 898, 173976. [Google Scholar] [CrossRef]
- Xu, S.; Feng, Y.; He, W.; Xu, W.; Xu, W.; Yang, H.; Li, X. Celastrol in metabolic diseases: Progress and application prospects. Pharmacol. Res. 2021, 167, 105572. [Google Scholar] [CrossRef]
- Abshagen, K.; Degenhardt, B.; Liebig, M.; Wendt, A.; Genz, B.; Schaeper, U.; Stumvoll, M.; Hofmann, U.; Frank, M.; Vollmar, B.; et al. Liver-specific Repin1 deficiency impairs transient hepatic steatosis in liver regeneration. Sci. Rep. 2018, 8, 16858. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Young-Cheul, K. Adipocyte differentiation and gene expression. J. Nutr. 2000, 130, 3122s–3126s. [Google Scholar] [CrossRef]
- Pan, S.; Yang, X.; Jia, Y.; Li, Y.; Chen, R.; Wang, M.; Cai, D.; Zhao, R. Intravenous injection of microvesicle-delivery miR-130b alleviates high-fat diet-induced obesity in C57BL/6 mice through translational repression of PPAR-γ. J. Biomed. Sci. 2015, 22, 86. [Google Scholar] [CrossRef]
- Uno, K.; Katagiri, H.; Yamada, T.; Ishigaki, Y.; Ogihara, T.; Imai, J.; Hasegawa, Y.; Gao, J.; Kaneko, K.; Iwasaki, H.; et al. Neuronal pathway from the liver modulates energy expenditure and systemic insulin sensitivity. Science 2006, 312, 1656–1659. [Google Scholar] [CrossRef]
- Korman, B.; Marangoni, R.G.; Lord, G.; Olefsky, J.; Tourtellotte, W.; Varga, J. Adipocyte-specific Repression of PPAR-gamma by NCoR Contributes to Scleroderma Skin Fibrosis. Arthritis Res. Ther. 2018, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jiang, C.; Kim, M.; Xiao, Y.; Richter, H.J.; Guan, D.; Zhu, K.; Krusen, B.M.; Roberts, A.N.; Miller, J.; et al. Isoform-specific functions of PPARγ in gene regulation and metabolism. Genes Dev. 2022, 36, 300–312. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) |
|---|---|
| H-PPARγ | Forward: GACCACTCCCACTCCTTTGA Reverse: CGACATTCAATTGCCATGAG |
| H-PPARγ1 | Forward: AGGCGAGGGCGATCTTGACAG Reverse: GATGCGGATGGCCACCTCTTT |
| H-PPARγ2 | Forward: GCCTTGCAGTGGGGATGTCTC Reverse: CCTGGGCGGTTGATTTGTCTG |
| H-PPARα | Forward: CTATCATTTGCTGTGGAGATCG Reverse: AAGATATCGTCCGGGTGGTT |
| H-CD36 | Forward: CATCGCTGGGGCTGTCATT Reverse: GCGTCCTGGGTTACATTTTCC |
| H-Fabp3 | Forward: TTCTGGAAGCTAGTGGACAG Reverse: TGATGGTAGTAGGCTTGGTCAT |
| H-Fatp1 | Forward: TGACAGTCGTCCTCCGCAAGAA Reverse: CTTCAGCAGGTAGCGGCAGATC |
| H-MCAD | Forward: TAATCGGTGAAGGAGCAGGTTT Reverse: GGCATACTTCGTGGCTTCGT |
| H-ACOX | Forward: GTTTGGACTCCGCCACTGCTTG Reverse: GGCTGAACTCTGGCATCCACAT |
| H-SCD1 | Forward: CAGGTTTCCAAGCGCAGTTC Reverse: ACTGGAGATCTCTTGGAGCA |
| H-Fads2 | Forward: TTCCTGGAGAGCCACTGGTTTG Reverse: GAAGAAGGACTGCTCCACATTGC |
| H-Acc1 | Forward: ATGGGCGGAATGGTCTCTTTC Reverse: TGGGGACCTTGTCTTCATCAT |
| H-Fasn | Forward: CACAGGGACAACCTGGAGTT Reverse: ACTCCACAGGTGGGAACAAG |
| H-Gapdh | Forward: ATGGATGATGATATCGCCGCC Reverse: CTCCATGTCGTCCAGTTGGT |
| M-PPARγ | Forward: TTTTCAAGGGTGCCAGTTTC Reverse: AATCCTTGGCCCTCTGAGAT |
| M-C/ebpα | Forward: CAAGAACAGCAACGAGTACCG Reverse: GTCACTGGTCAACTCCAGCAC |
| M-aP2 | Forward: AGGTGAAGAGCATCATAACCCT Reverse: TCACGCCTTTCATAACACATTCC |
| M-Adipoq | Forward: TGTTCCTCTTAATCCTGCCCA Reverse: CCAACCTGCACAAGTTCCCTT |
| M-LPL | Forward: GGGAGTTTGGCTCCAGAGTTT Reverse: TGTGTCTTCAGGGGTCCTTAG |
| M-Pgc-1α | Forward: ACCCACAGGATCAGAACAAACCCT Reverse: TTGGTGTGAGGAGGGTCATCGTTT |
| M-Atgl | Forward: GTTGAAGGAGGGATGCAGAG Reverse: GCCACTCACATCTACGGAGC |
| M-Lipe | Forward: TCTCGTTGCGTTTGTAGTGC Reverse: ACGCTACACAAAGGCTGCTT |
| M-Mgl | Forward: AACCACTAAGCCCCAGTTCC Reverse: GCAGGATGTGAGCAGAGCAC |
| M-Gapdh | Forward: AACTTTGGCATTGTGGAAGG Reverse: ACACATTGGGGGTAGGAACAM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, M.; Wang, Y.; Ma, Y.; Li, J.; Wang, J.; Liu, C. Celastrol Stabilizes Glycolipid Metabolism in Hepatic Steatosis by Binding and Regulating the Peroxisome Proliferator-Activated Receptor γ Signaling Pathway. Metabolites 2024, 14, 64. https://doi.org/10.3390/metabo14010064
Luo M, Wang Y, Ma Y, Li J, Wang J, Liu C. Celastrol Stabilizes Glycolipid Metabolism in Hepatic Steatosis by Binding and Regulating the Peroxisome Proliferator-Activated Receptor γ Signaling Pathway. Metabolites. 2024; 14(1):64. https://doi.org/10.3390/metabo14010064
Chicago/Turabian StyleLuo, Mingzhu, Yiting Wang, Yanyan Ma, Jingzhe Li, Jingyi Wang, and Changzhen Liu. 2024. "Celastrol Stabilizes Glycolipid Metabolism in Hepatic Steatosis by Binding and Regulating the Peroxisome Proliferator-Activated Receptor γ Signaling Pathway" Metabolites 14, no. 1: 64. https://doi.org/10.3390/metabo14010064
APA StyleLuo, M., Wang, Y., Ma, Y., Li, J., Wang, J., & Liu, C. (2024). Celastrol Stabilizes Glycolipid Metabolism in Hepatic Steatosis by Binding and Regulating the Peroxisome Proliferator-Activated Receptor γ Signaling Pathway. Metabolites, 14(1), 64. https://doi.org/10.3390/metabo14010064