
Unveiling the Nexus: Cellular Metabolomics Unravels the Impact of Estrogen on Nicotinamide Metabolism in Mitigating Rheumatoid Arthritis Pathogenesis


Abstract
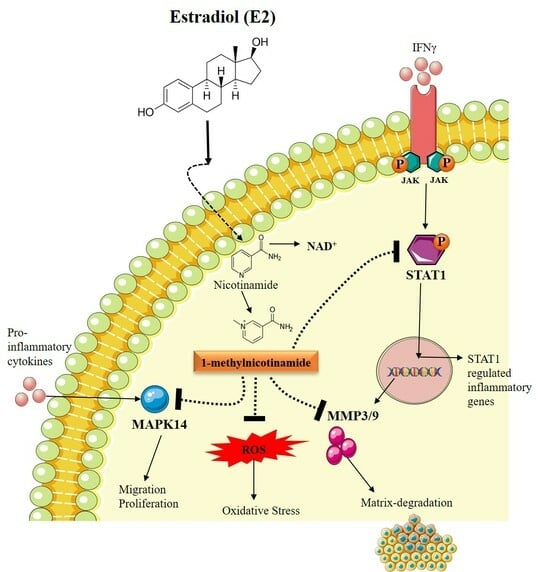
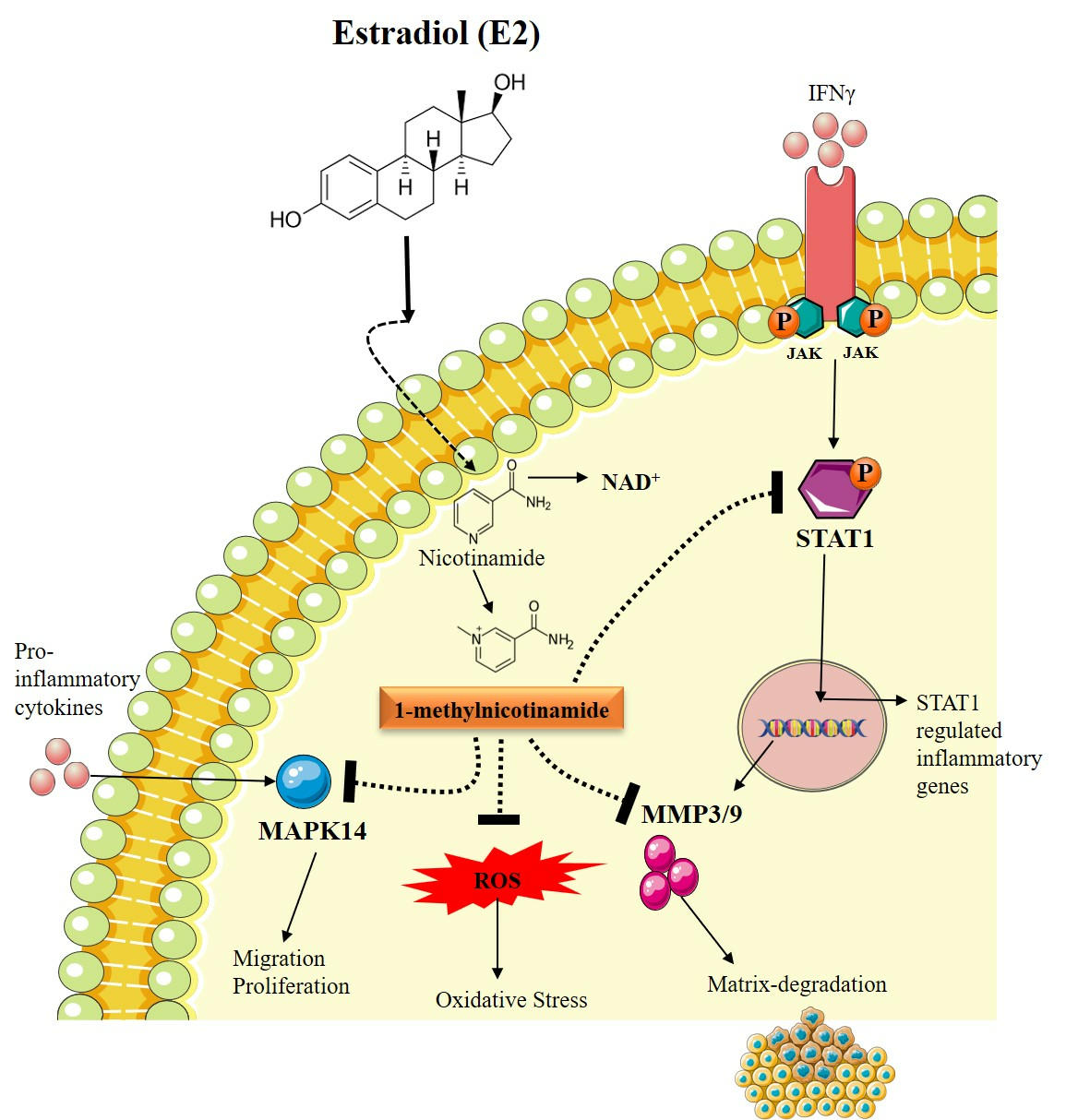
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Clinical Sample Collection
2.2. Cell Culture and Treatment
2.3. Metabolite Extraction
2.4. Data Processing
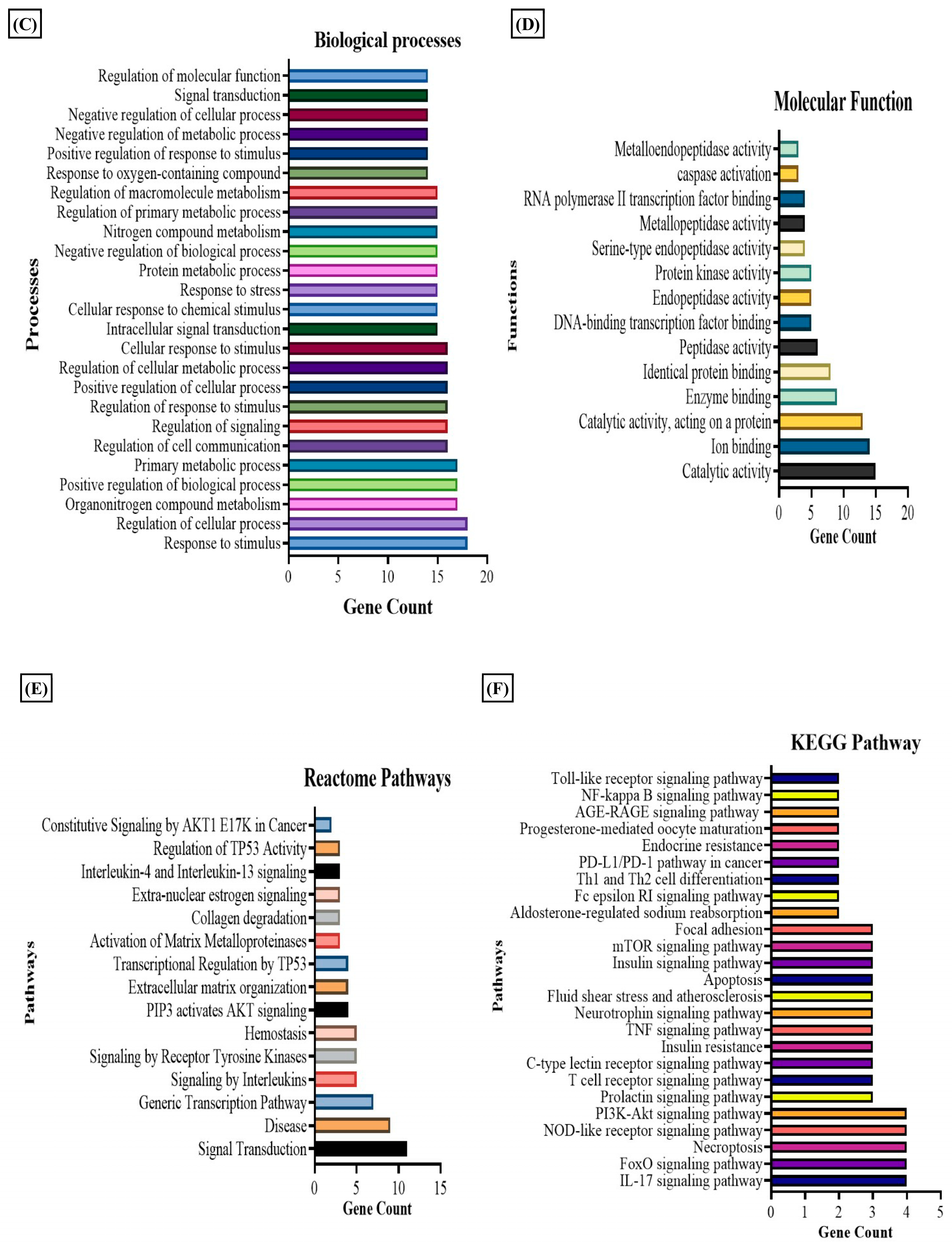
2.5. Target Prediction of Metabolites by PharmMapper Analysis
2.6. RNA Isolation and qRT-PCR
2.7. Western Blotting
2.8. Detection of Total Cellular ROS Production
2.9. Statistical Analysis
3. Results
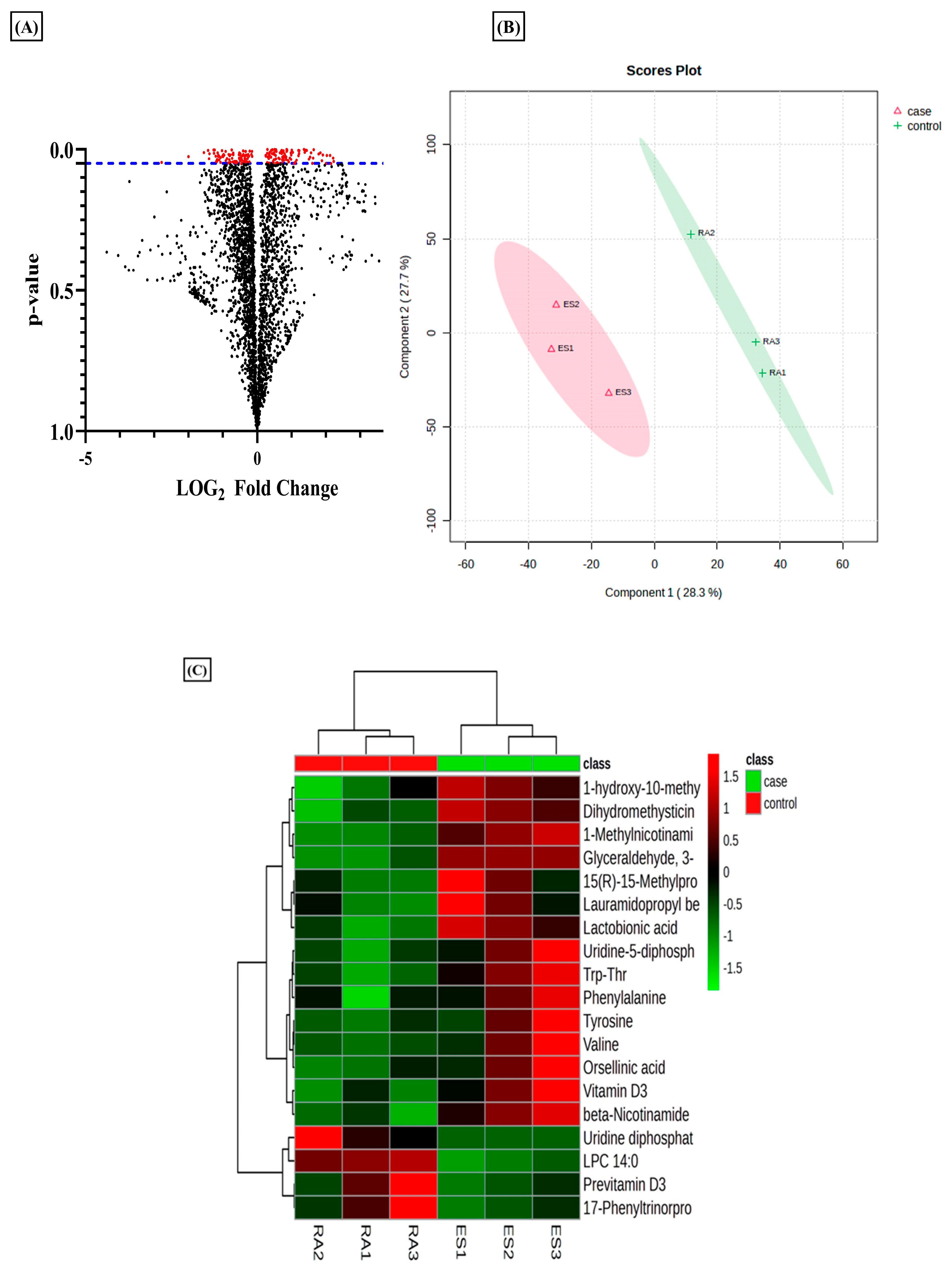
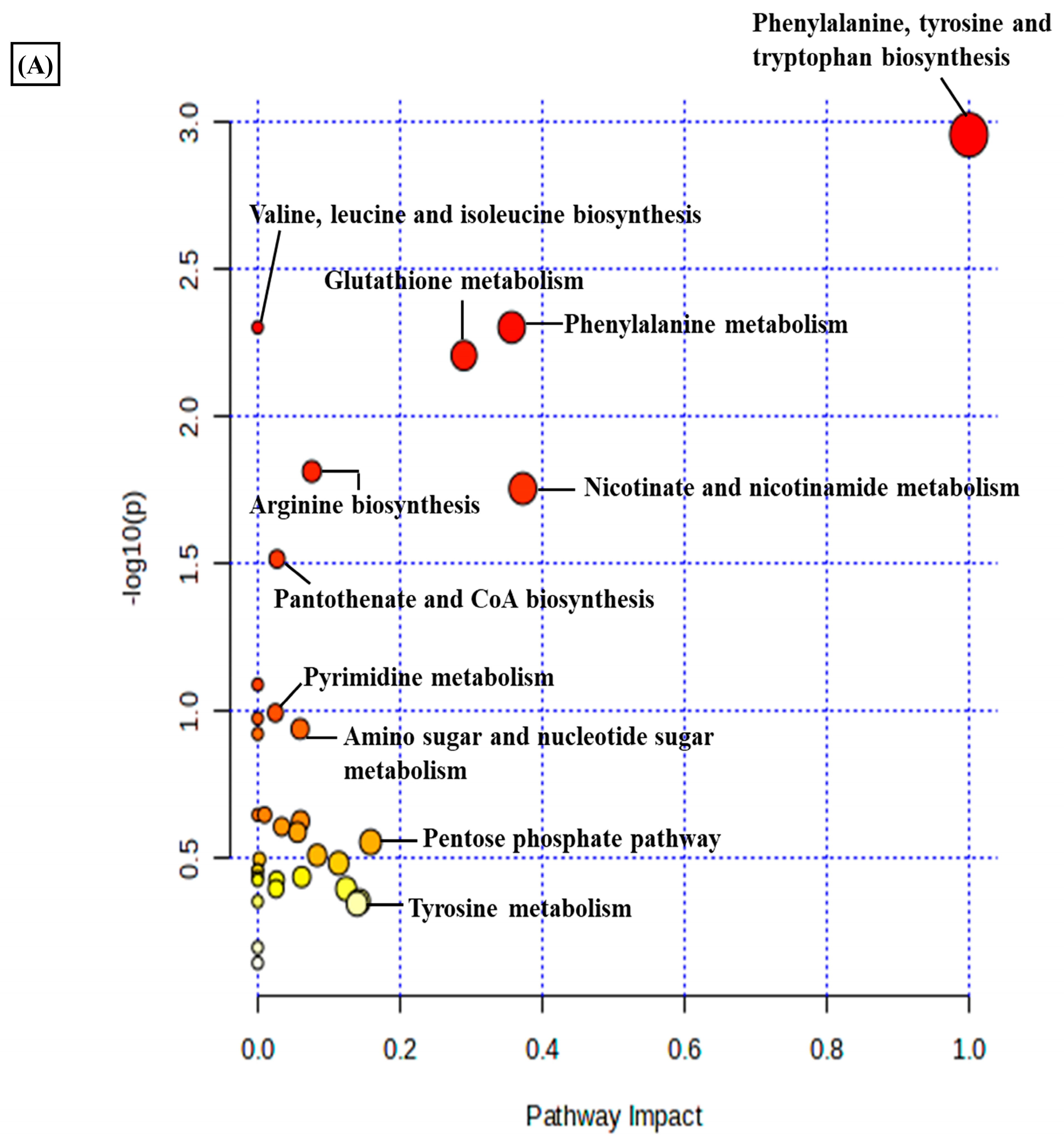
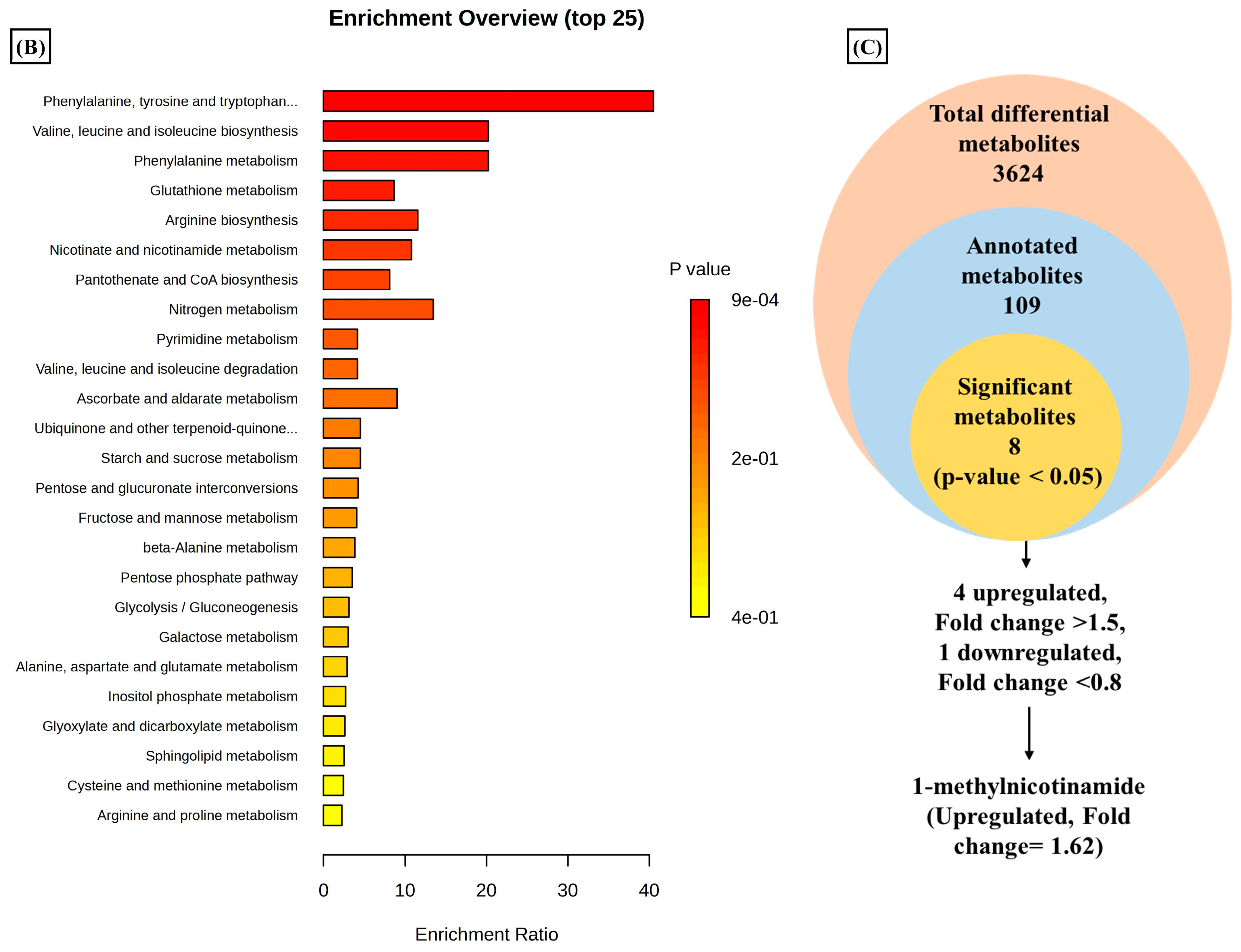
3.1. Differential Metabolomic Analysis of RA Synovial Fibroblast upon Estradiol Induction
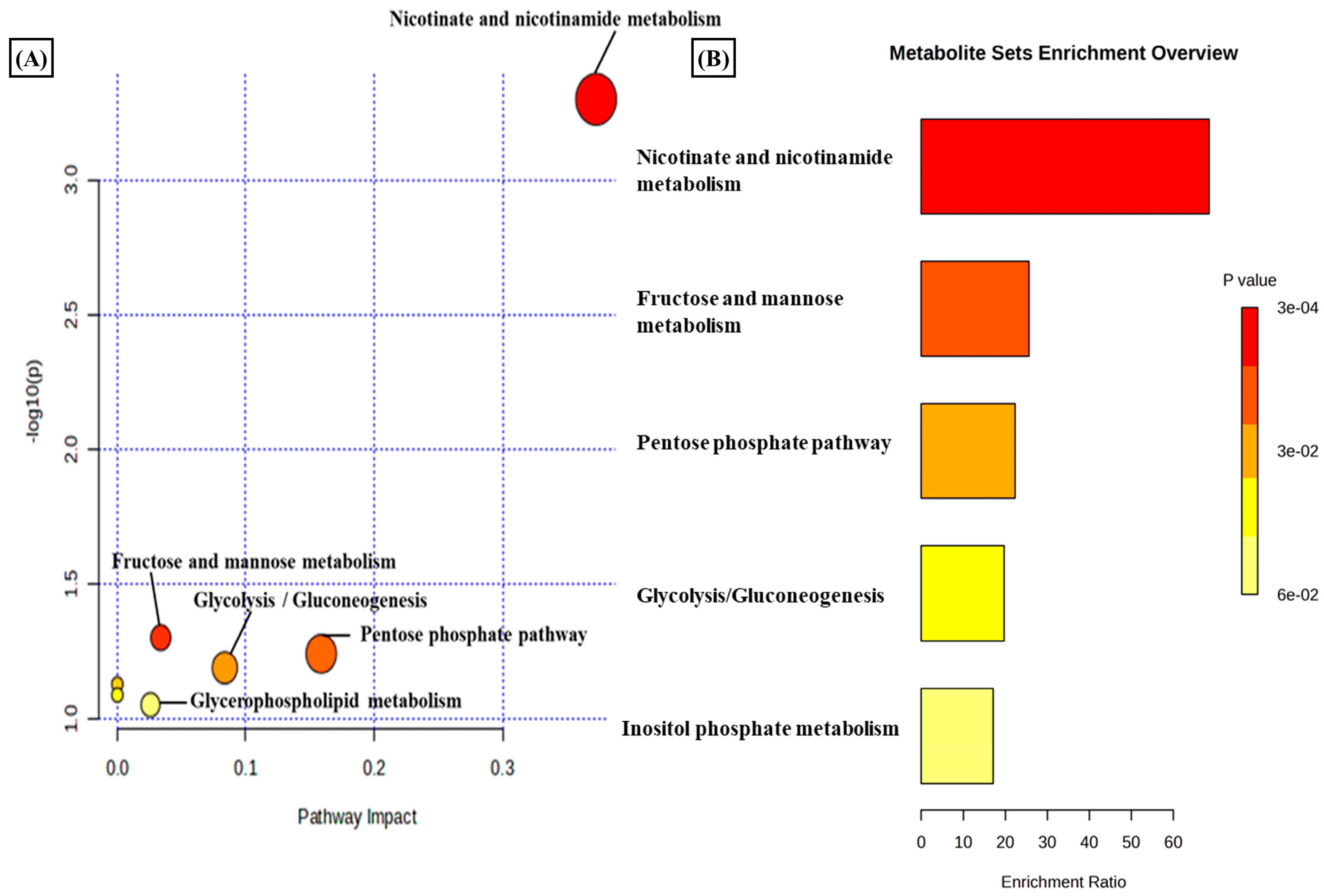
3.2. Estradiol Promotes Nicotinamide Metabolism in RA Synovial Fibroblasts
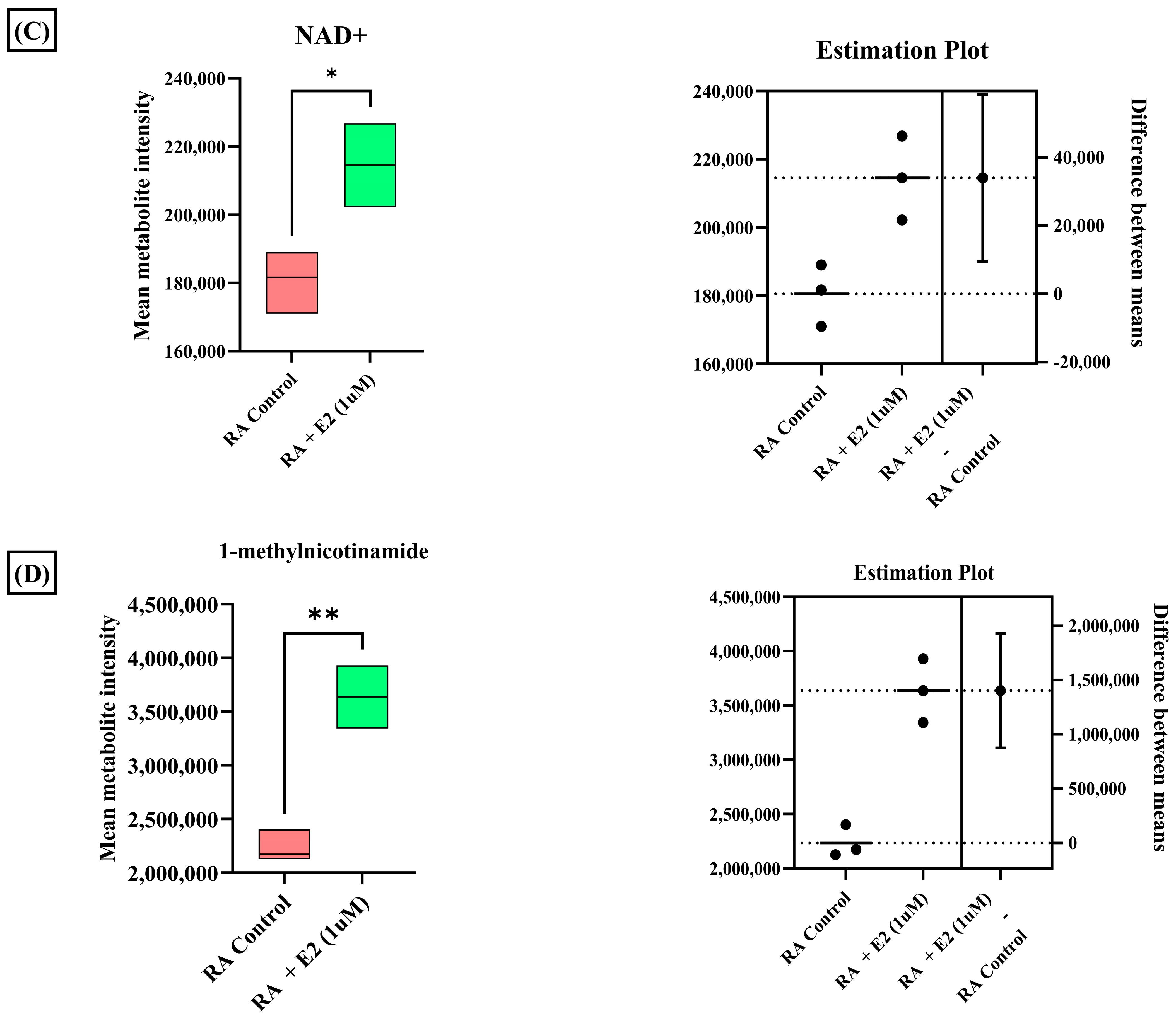
3.3. Estradiol Affects RA Pathogenesis by Stimulating 1-Methyl Nicotinamide
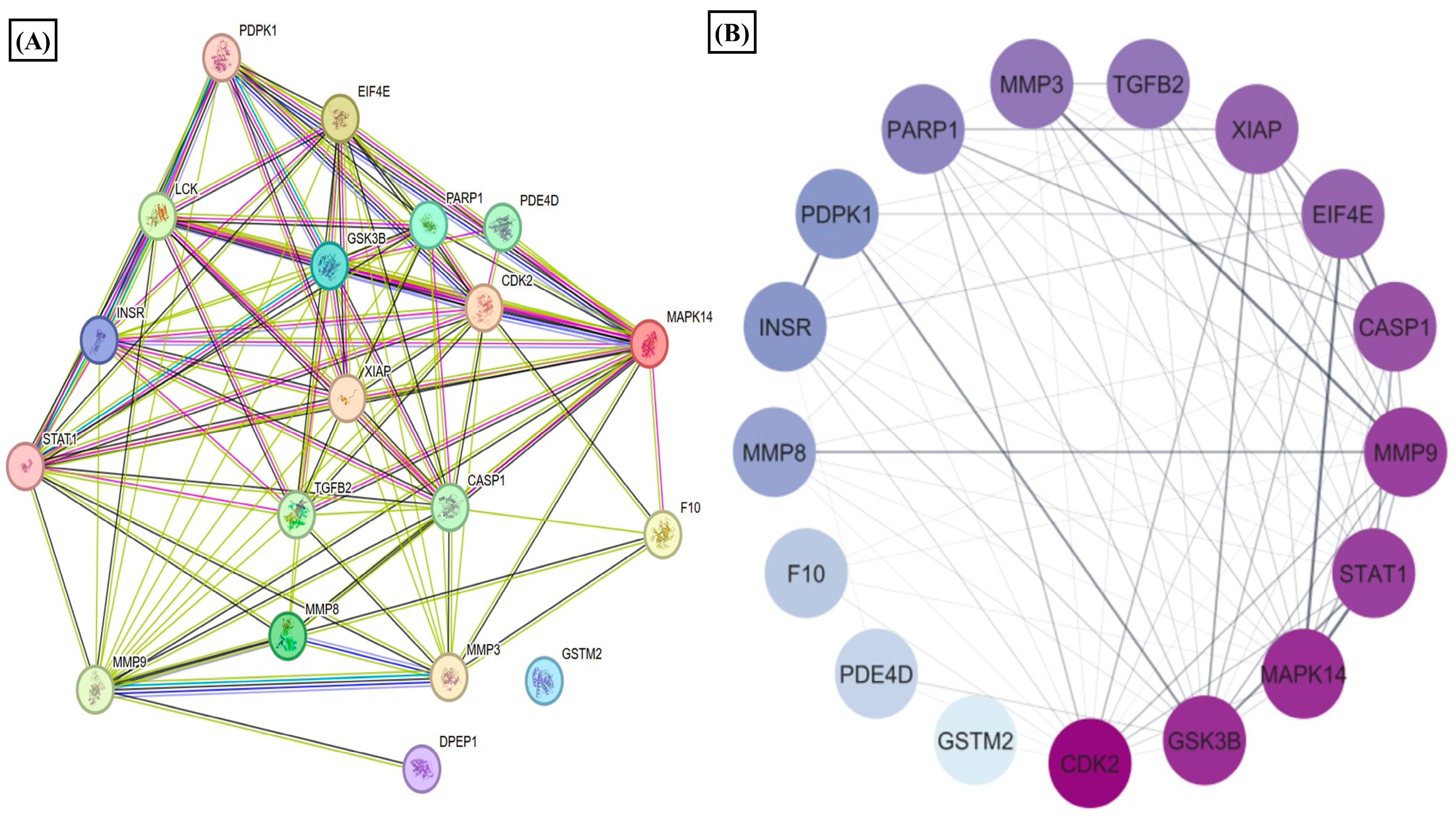
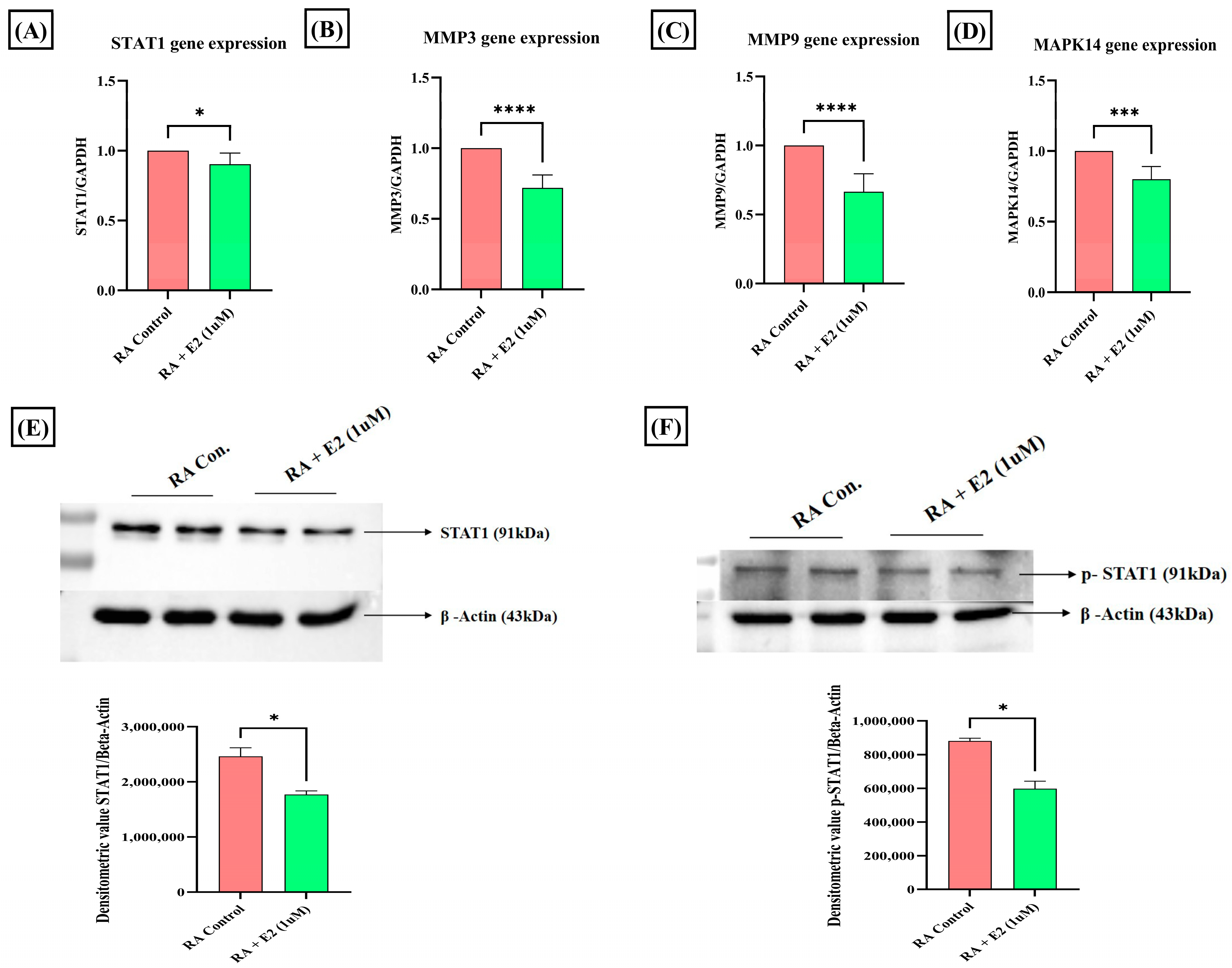
3.4. Estradiol Alters STAT1 Signaling in RA Synovial Fibroblast through 1-Methylnicotinamide
3.5. Estradiol Downregulates Matrix-Degrading Enzymes and MAPK14 Expression in RA-FLS Mediated by 1-Methylnicotinamide
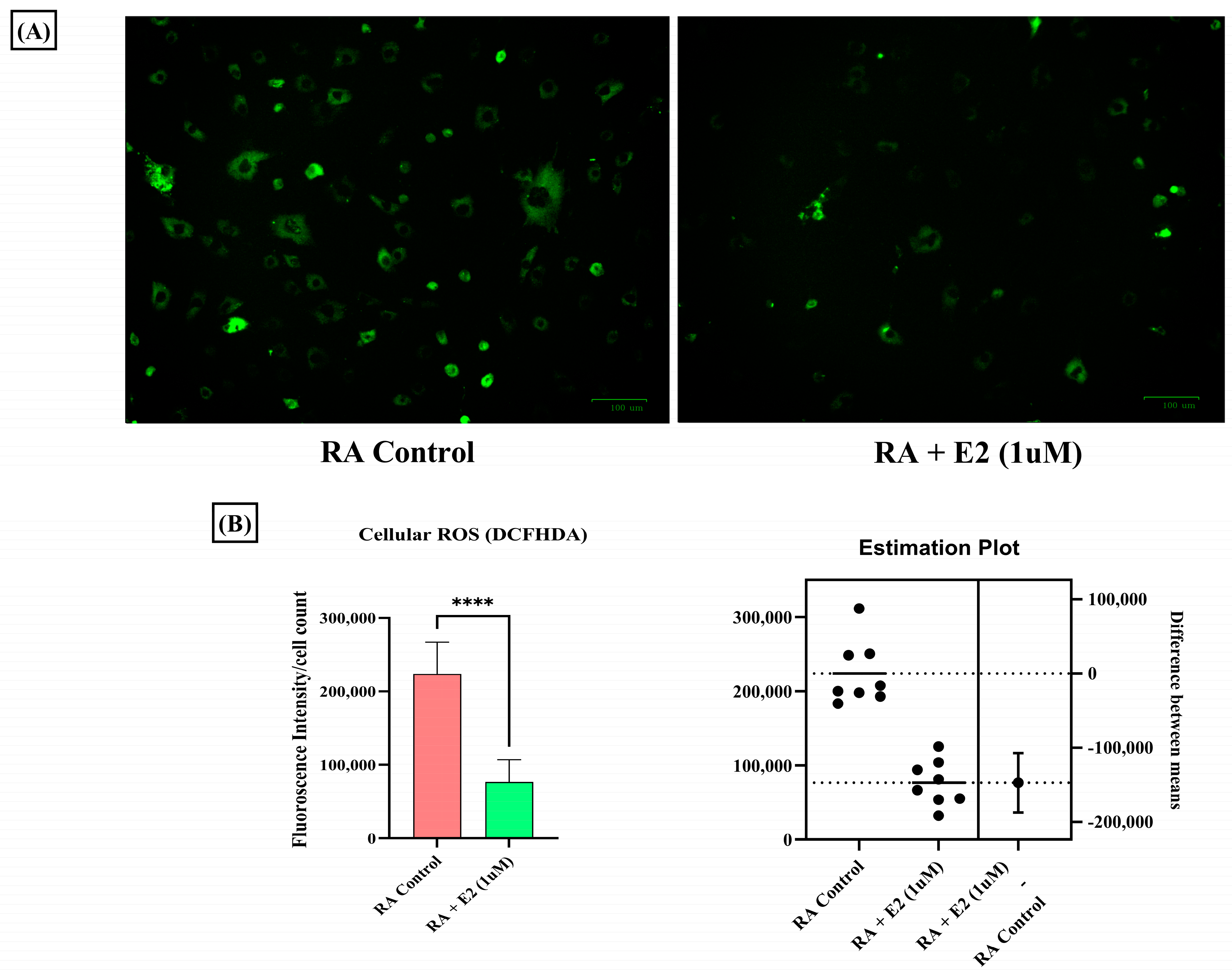
3.6. Estradiol Ameliorates ROS Production in RA-FLS Regulated by 1-MNA Generation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chimenti, M.S.; Triggianese, P.; Conigliaro, P.; Candi, E.; Melino, G.; Perricone, R. The Interplay between Inflammation and Metabolism in Rheumatoid Arthritis. Cell Death Dis. 2015, 6, e1887. [Google Scholar] [CrossRef] [PubMed]
- Slobodin, G. Rheumatoid Arthritis BT. In Rheumatic Disease in Geriatrics: Diagnosis and Management; Slobodin, G., Shoenfeld, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 173–183. ISBN 978-3-030-44234-7. [Google Scholar]
- Black, R.J.; Cross, M.; Haile, L.M.; Culbreth, G.T.; Steinmetz, J.D.; Hagins, H.; Kopec, J.A.; Brooks, P.M.; Woolf, A.D.; Ong, K.L.; et al. Global, Regional, and National Burden of Rheumatoid Arthritis, 1990–2020, and Projections to 2050: A Systematic Analysis of the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e594–e610. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Pope, J.E. Novel Treatment Strategies in Rheumatoid Arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Breedveld, F.C.; Burmester, G.R.; Bykerk, V.; Dougados, M.; Emery, P.; Kvien, T.K.; Navarro-Compán, M.V.; Oliver, S.; Schoels, M.; et al. Treating Rheumatoid Arthritis to Target: 2014 Update of the Recommendations of an International Task Force. Ann. Rheum. Dis. 2016, 75, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, E.; Adami, G.; Rossini, M.; Giollo, A.; Orsolini, G.; Viapiana, O.; Gatti, D.; Fassio, A. The Emerging Roles of Endocrine Hormones in Different Arthritic Disorders. Front. Endocrinol. 2021, 12, 620920. [Google Scholar] [CrossRef] [PubMed]
- Kvien, T.K.; Uhlig, T.; Ødegård, S.; Heiberg, M.S. Epidemiological Aspects of Rheumatoid Arthritis: The Sex Ratio. Ann. N. Y. Acad. Sci. 2006, 1069, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, K.; Jandu, J.S.; Brent, L.H.; Al-Dhahir, M.A. Rheumatoid Arthritis; NIH: Treasure Island, FL, USA, 2024. [Google Scholar]
- Wong, L.E.; Huang, W.-T.; Pope, J.E.; Haraoui, B.; Boire, G.; Thorne, J.C.; Hitchon, C.A.; Tin, D.; Keystone, E.C.; Bykerk, V.P. Effect of Age at Menopause on Disease Presentation in Early Rheumatoid Arthritis: Results from the Canadian Early Arthritis Cohort. Arthritis Care Res. 2015, 67, 616–623. [Google Scholar] [CrossRef]
- Gold, S.M.; Sasidhar, M.V.; Morales, L.B.; Du, S.; Sicotte, N.L.; Tiwari-Woodruff, S.K.; Voskuhl, R.R. Estrogen Treatment Decreases Matrix Metalloproteinase (MMP)-9 in Autoimmune Demyelinating Disease through Estrogen Receptor Alpha (ERα). Lab. Investig. 2009, 89, 1076–1083. [Google Scholar] [CrossRef]
- Ostensen, M.; Aune, B.; Husby, G. Effect of Pregnancy and Hormonal Changes on the Activity of Rheumatoid Arthritis. Scand. J. Rheumatol. 1983, 12, 69–72. [Google Scholar] [CrossRef]
- Kanik, K.S.; Wilder, R.L. Hormonal Alterations in Rheumatoid Arthritis, including the Effects of Pregnancy. Rheum. Dis. Clin. N. Am. 2000, 26, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, D.; Enokida, M.; Okano, T.; Hagino, H.; Teshima, R. Effects of Ovariectomy and Estrogen Replacement Therapy on Arthritis and Bone Mineral Density in Rats with Collagen-Induced Arthritis. Bone 2001, 28, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Jansson, L.; Holmdahl, R. Enhancement of Collagen-Induced Arthritis in Female Mice by Estrogen Receptor Blockage. Arthritis Rheum. 2001, 44, 2168–2175. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Balachandran, C.; Manohar, B.M.; Puvanakrishnan, R. Effects of Testosterone, Estrogen and Progesterone on TNF-α Mediated Cellular Damage in Rat Arthritic Synovial Fibroblasts. Rheumatol. Int. 2012, 32, 3181–3188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, M. [Corrigendum] Estrogen Downregulates TAK1 Expression in Human Fibroblast-like Synoviocytes and in a Rheumatoid Arthritis Model. Exp. Ther. Med. 2022, 23, 225. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, C.; Bondt, A.; Harre, U.; Raufer, J.; Pfeifle, R.; Camponeschi, A.; Wuhrer, M.; Seeling, M.; Mårtensson, I.-L.; Nimmerjahn, F.; et al. Estrogen Induces St6gal1 Expression and Increases IgG Sialylation in Mice and Patients with Rheumatoid Arthritis: A Potential Explanation for the Increased Risk of Rheumatoid Arthritis in Postmenopausal Women. Arthritis Res. Ther. 2018, 20, 84. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Sarkar, A.; Mann, S.; Monu, M.; Agnihotri, P.; Saquib, M.; Malik, S.; Kumavat, R.; Mathur, A.; Biswas, S. Estrogen-Mediated Differential Protein Regulation and Signal Transduction in Rheumatoid Arthritis. J. Mol. Endocrinol. 2022, 69, R25–R43. [Google Scholar] [CrossRef]
- Hanlon, M.M.; Canavan, M.; Barker, B.E.; Fearon, U. Metabolites as Drivers and Targets in Rheumatoid Arthritis. Clin. Exp. Immunol. 2022, 208, 167–180. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, J.; Hu, C.; Xie, Z.; Li, H.; Wei, S.; Wang, D.; Wen, C.; Xu, G. Exploration of the Serum Metabolite Signature in Patients with Rheumatoid Arthritis Using Gas Chromatography–Mass Spectrometry. J. Pharm. Biomed. Anal. 2016, 127, 60–67. [Google Scholar] [CrossRef]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 Plays a Key Role in the Cell Growth of Fibroblast-like Synoviocytes in Rheumatoid Arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef]
- Huffman, K.M.; Jessee, R.; Andonian, B.; Davis, B.N.; Narowski, R.; Huebner, J.L.; Kraus, V.B.; McCracken, J.; Gilmore, B.F.; Tune, K.N.; et al. Molecular Alterations in Skeletal Muscle in Rheumatoid Arthritis Are Related to Disease Activity, Physical Inactivity, and Disability. Arthritis Res. Ther. 2017, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, X.; Wang, W.; Peng, Y.; Bi, K.; Dai, R. Targeted Profiling of Arachidonic Acid and Eicosanoids in Rat Tissue by UFLC–MS/MS: Application to Identify Potential Markers for Rheumatoid Arthritis. Talanta 2017, 162, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhu, Y.; Zhuge, T.; Li, B.; Gu, C. Metabolomics Analysis Discovers Estrogen Altering Cell Proliferation via the Pentose Phosphate Pathway in Infertility Patient Endometria. Front. Endocrinol. 2021, 12, 791174. [Google Scholar] [CrossRef] [PubMed]
- Clish, C.B. Metabolomics: An Emerging but Powerful Tool for Precision Medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Kuehnbaum, N.L.; Britz-McKibbin, P. New Advances in Separation Science for Metabolomics: Resolving Chemical Diversity in a Post-Genomic Era. Chem. Rev. 2013, 113, 2437–2468. [Google Scholar] [CrossRef]
- Kim, S.; Hwang, J.; Xuan, J.; Jung, Y.H.; Cha, H.-S.; Kim, K.H. Global Metabolite Profiling of Synovial Fluid for the Specific Diagnosis of Rheumatoid Arthritis from Other Inflammatory Arthritis. PLoS ONE 2014, 9, e97501. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, P.; Monu; Ramani, S.; Chakraborty, D.; Saquib, M.; Biswas, S. Differential Metabolome in Rheumatoid Arthritis: A Brief Perspective. Curr. Rheumatol. Rep. 2021, 23, 42. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Harms, A.C.; van Wijk, E.; Wang, M.; Berger, R.; Koval, S.; Hankemeier, T.; van der Greef, J. Role of Amino Acids in Rheumatoid Arthritis Studied by Metabolomics. Int. J. Rheum. Dis. 2019, 22, 38–46. [Google Scholar] [CrossRef]
- Li, J.; Che, N.; Xu, L.; Zhang, Q.; Wang, Q.; Tan, W.; Zhang, M. LC-MS-Based Serum Metabolomics Reveals a Distinctive Signature in Patients with Rheumatoid Arthritis. Clin. Rheumatol. 2018, 37, 1493–1502. [Google Scholar] [CrossRef]
- Tatar, Z.; Migne, C.; Petera, M.; Gaudin, P.; Lequerre, T.; Marotte, H.; Tebib, J.; Pujos Guillot, E.; Soubrier, M. Variations in the Metabolome in Response to Disease Activity of Rheumatoid Arthritis. BMC Musculoskelet. Disord. 2016, 17, 353. [Google Scholar] [CrossRef]
- Wise, P.M.; Suzuki, S.; Brown, C.M. Estradiol: A Hormone with Diverse and Contradictory Neuroprotective Actions. Dialogues Clin. Neurosci. 2009, 11, 297–303. [Google Scholar] [CrossRef]
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite Identification and Quantitation in LC-MS/MS-Based Metabolomics. Trends Anal. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef]
- Monu; Agnihotri, P.; Saquib, M.; Sarkar, A.; Chakraborty, D.; Kumar, U.; Biswas, S. Transthyretin and Receptor for Advanced Glycation End Product’s Differential Levels Associated with the Pathogenesis of Rheumatoid Arthritis. J. Inflamm. Res. 2021, 14, 5581–5596. [Google Scholar] [CrossRef]
- Tanaka, I.; Morikawa, M.; Okuse, T.; Shirakawa, M.; Imai, K. Expression and Regulation of WISP2 in Rheumatoid Arthritic Synovium. Biochem. Biophys. Res. Commun. 2005, 334, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Berthois, Y.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Phenol Red in Tissue Culture Media Is a Weak Estrogen: Implications Concerning the Study of Estrogen-Responsive Cells in Culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, M.; Xu, L.; Shi, Z. A Reversed-Phase/Hydrophilic Interaction Mixed-Mode C18-Diol Stationary Phase for Multiple Applications. Anal. Chim. Acta 2015, 888, 182–190. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper Server: A Web Server for Potential Drug Target Identification Using Pharmacophore Mapping Approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef]
- Piñero, J.; Queralt-Rosinach, N.; Bravo, À.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A Discovery Platform for the Dynamical Exploration of Human Diseases and Their Genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Chakraborty, D.; Tiwari, A.; Biswas, S. In Silico Studies on the Phytochemical Components of Murraya Koenigii Targeting TNF-α in Rheumatoid Arthritis. J. Herb. Med. 2020, 24, 100396. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Sharma, S.; Agnihotri, P.; Sarkar, T.; Kumari, P.; Malhotra, R.; Datta, B.; Kumar, V.; Biswas, S. Synovial Fluid Cell Proteomic Analysis Identifies Upregulation of Alpha-Taxilin Proteins in Rheumatoid Arthritis: A Potential Prognostic Marker. J. Immunol. Res. 2020, 2020, 4897983. [Google Scholar] [CrossRef]
- Stanford, S.M.; Maestre, M.F.; Campbell, A.M.; Bartok, B.; Kiosses, W.B.; Boyle, D.L.; Arnett, H.A.; Mustelin, T.; Firestein, G.S.; Bottini, N. Protein Tyrosine Phosphatase Expression Profile of Rheumatoid Arthritis Fibroblast-like Synoviocytes: A Novel Role of SH2 Domain-Containing Phosphatase 2 as a Modulator of Invasion and Survival. Arthritis Rheum. 2013, 65, 1171–1180. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS Using Oxidized DCFDA and Flow-Cytometry. In Advanced Protocols in Oxidative Stress II; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 57–72. ISBN 978-1-60761-411-1. [Google Scholar]
- Kim, H.; Xue, X. Detection of Total Reactive Oxygen Species in Adherent Cells by 2’,7’-Dichlorodihydrofluorescein Diacetate Staining. J. Vis. Exp. 2020, 160. [Google Scholar] [CrossRef]
- Al-Azab, M.; Qaed, E.; Ouyang, X.; Elkhider, A.; Walana, W.; Li, H.; Li, W.; Tang, Y.; Adlat, S.; Wei, J.; et al. TL1A/TNFR2-Mediated Mitochondrial Dysfunction of Fibroblast-like Synoviocytes Increases Inflammatory Response in Patients with Rheumatoid Arthritis via Reactive Oxygen Species Generation. FEBS J. 2020, 287, 3088–3104. [Google Scholar] [CrossRef] [PubMed]
- Amruta, N.; Bix, G. ATN-161 Ameliorates Ischemia/Reperfusion-Induced Oxidative Stress, Fibro-Inflammation, Mitochondrial Damage, and Apoptosis-Mediated Tight Junction Disruption in BEnd.3 Cells. Inflammation 2021, 44, 2377–2394. [Google Scholar] [CrossRef] [PubMed]
- Blazejczyk, A.; Switalska, M.; Chlopicki, S.; Marcinek, A.; Gebicki, J.; Nowak, M.; Nasulewicz-Goldeman, A.; Wietrzyk, J. 1-Methylnicotinamide and Its Structural Analog 1,4-Dimethylpyridine for the Prevention of Cancer Metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 110. [Google Scholar] [CrossRef]
- Li, L.; Freitag, J.; Asbrand, C.; Munteanu, B.; Wang, B.-T.; Zezina, E.; Didier, M.; Thill, G.; Rocher, C.; Herrmann, M.; et al. Multi-Omics Profiling of Collagen-Induced Arthritis Mouse Model Reveals Early Metabolic Dysregulation via SIRT1 Axis. Sci. Rep. 2022, 12, 11830. [Google Scholar] [CrossRef]
- Kasperkovitz, P.V.; Verbeet, N.L.; Smeets, T.J.; van Rietschoten, J.G.I.; Kraan, M.C.; van der Pouw Kraan, T.C.T.M.; Tak, P.P.; Verweij, C.L. Activation of the STAT1 Pathway in Rheumatoid Arthritis. Ann. Rheum. Dis. 2004, 63, 233–239. [Google Scholar] [CrossRef]
- Balendran, T.; Lim, K.; Hamilton, J.A.; Achuthan, A.A. Targeting Transcription Factors for Therapeutic Benefit in Rheumatoid Arthritis. Front. Immunol. 2023, 14, 1196931. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Transcription Factor Activity of STAT Proteins: Structural Requirements and Regulation by Phosphorylation and Interacting Proteins. Cell Mol. Life Sci. 1999, 55, 1535–1546. [Google Scholar] [CrossRef]
- Fiedorczyk, M.; Klimiuk, P.A.; Sierakowski, S.; Domysławska, I.; Chwiećko, J. Correlations between serum matrix metalloproteinase (MMP-1, MMP-3, MMP-9, MMP-13) concentrations and markers of disease activity in early rheumatoid arthritis. Przegl. Lek. 2005, 62, 1321–1324. [Google Scholar]
- Bian, Y.; Xiang, Z.; Wang, Y.; Ren, Q.; Chen, G.; Xiang, B.; Wang, J.; Zhang, C.; Pei, S.; Guo, S.; et al. Immunomodulatory Roles of Metalloproteinases in Rheumatoid Arthritis. Front. Pharmacol. 2023, 14, 1285455. [Google Scholar] [CrossRef]
- Limoge, M.; Safina, A.; Truskinovsky, A.M.; Aljahdali, I.; Zonneville, J.; Gruevski, A.; Arteaga, C.L.; Bakin, A.V. Tumor P38MAPK Signaling Enhances Breast Carcinoma Vascularization and Growth by Promoting Expression and Deposition of Pro-Tumorigenic Factors. Oncotarget 2017, 8, 61969–61981. [Google Scholar] [CrossRef]
- Böhm, C.; Hayer, S.; Kilian, A.; Zaiss, M.M.; Finger, S.; Hess, A.; Engelke, K.; Kollias, G.; Krönke, G.; Zwerina, J.; et al. The α-Isoform of P38 MAPK Specifically Regulates Arthritic Bone Loss1. J. Immunol. 2009, 183, 5938–5947. [Google Scholar] [CrossRef]
- Sidor, K.; Jeznach, A.; Hoser, G.; Skirecki, T. 1-Methylnicotinamide (1-MNA) Inhibits the Activation of the NLRP3 Inflammasome in Human Macrophages. Int. Immunopharmacol. 2023, 121, 110445. [Google Scholar] [CrossRef]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Cheng, A.; Guma, M. Can Metabolic Pathways Be Therapeutic Targets in Rheumatoid Arthritis? J. Clin. Med. 2019, 8, 753. [Google Scholar] [CrossRef]
- Lefevre, S.; Meier, F.M.P.; Neumann, E.; Muller-Ladner, U. Role of Synovial Fibroblasts in Rheumatoid Arthritis. Curr. Pharm. Des. 2015, 21, 130–141. [Google Scholar] [CrossRef]
- José Alcaraz, M. New Potential Therapeutic Approaches Targeting Synovial Fibroblasts in Rheumatoid Arthritis. Biochem. Pharmacol. 2021, 194, 114815. [Google Scholar] [CrossRef] [PubMed]
- de Jong, T.A.; Semmelink, J.F.; Denis, S.W.; van de Sande, M.G.H.; Houtkooper, R.H.L.; van Baarsen, L.G.M. Altered Lipid Metabolism in Synovial Fibroblasts of Individuals at Risk of Developing Rheumatoid Arthritis. J. Autoimmun. 2023, 134, 102974. [Google Scholar] [CrossRef] [PubMed]
- Aghakhani, S.; Zerrouk, N.; Niarakis, A. Metabolic Reprogramming of Fibroblasts as Therapeutic Target in Rheumatoid Arthritis and Cancer: Deciphering Key Mechanisms Using Computational Systems Biology Approaches. Cancers 2021, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.T. Hormonal and Immunologic Risk Factors for the Development of Rheumatoid Arthritis: An Integrative Physiopathogenetic Perspective. Rheum. Dis. Clin. N. Am. 2000, 26, 775–803. [Google Scholar] [CrossRef] [PubMed]
- Romo-García, M.F.; Zapata-Zuñiga, M.; Enciso-Moreno, J.A.; Castañeda-Delgado, J.E. The Role of Estrogens. In Rheumatoid Arthritis Physiopathology; Mohammed, R.H.A., Ed.; IntechOpen: Rijeka, Croatia, 2020; Chapter 2; ISBN 978-1-83962-533-6. [Google Scholar]
- Mitani, M.; Miura, Y.; Saura, R.; Kitagawa, A.; Fukuyama, T.; Hashiramoto, A.; Shiozawa, S.; Kurosaka, M.; Yoshiya, S. Estrogen Specifically Stimulates Expression and Production of Osteoprotegerin from Rheumatoid Synovial Fibroblasts. Int. J. Mol. Med. 2005, 15, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sanchez, C.; Escudero-Contreras, A.; Cerdó, T.; Sánchez-Mendoza, L.M.; Llamas-Urbano, A.; la Rosa, I.A.; Pérez-Rodriguez, M.; Muñoz-Barrera, L.; Del Carmen Abalos-Aguilera, M.; Barbarroja, N.; et al. Preclinical Characterization of Pharmacologic NAD(+) Boosting as a Promising Therapeutic Approach in Rheumatoid Arthritis. Arthritis Rheumatol. 2023, 75, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a Primary Metabolite of Nicotinamide, Exerts Anti-Thrombotic Activity Mediated by a Cyclooxygenase-2/Prostacyclin Pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Gebicki, J.; Sysa-Jedrzejowska, A.; Adamus, J.; Woźniacka, A.; Rybak, M.; Zielonka, J. 1-Methylnicotinamide: A Potent Anti-Inflammatory Agent of Vitamin Origin. Pol. J. Pharmacol. 2003, 55, 109–112. [Google Scholar] [PubMed]
- Wozniacka, A.; Wieczorkowska, M.; Gebicki, J.; Sysa-Jedrzejowska, A. Topical Application of 1-methylnicotinamide in the Treatment of Rosacea: A Pilot Study. Clin. Exp. Dermatol. 2005, 30, 632–635. [Google Scholar] [CrossRef]
- Wang, X.; Pan, C.; Gong, J.; Liu, X.; Li, H. Enhancing the Enrichment of Pharmacophore-Based Target Prediction for the Polypharmacological Profiles of Drugs. J. Chem. Inf. Model. 2016, 56, 1175–1183. [Google Scholar] [CrossRef]
- Choy, E. Understanding the Dynamics: Pathways Involved in the Pathogenesis of Rheumatoid Arthritis. Rheumatology 2012, 51, v3–v11. [Google Scholar] [CrossRef]
- Ramana, C.V.; Chatterjee-Kishore, M.; Nguyen, H.; Stark, G.R. Complex Roles of Stat1 in Regulating Gene Expression. Oncogene 2000, 19, 2619–2627. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Wu, C.; Sun, S.; Pan, J. E2F2 Directly Regulates the STAT1 and PI3K/AKT/NF-ΚB Pathways to Exacerbate the Inflammatory Phenotype in Rheumatoid Arthritis Synovial Fibroblasts and Mouse Embryonic Fibroblasts. Arthritis Res. Ther. 2018, 20, 225. [Google Scholar] [CrossRef] [PubMed]
- Taniki, N.; Nakamoto, N.; Chu, P.-S.; Mikami, Y.; Amiya, T.; Teratani, T.; Suzuki, T.; Tsukimi, T.; Fukuda, S.; Yamaguchi, A.; et al. Intestinal Barrier Regulates Immune Responses in the Liver via IL-10-Producing Macrophages. JCI Insight 2018, 3, e91980. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Tucci, G.; Garufi, C.; Pacella, I.; Zagaglioni, M.; Pinzon Grimaldos, A.; Ceccarelli, F.; Conti, F.; Spinelli, F.R.; Piconese, S. Baricitinib Therapy Response in Rheumatoid Arthritis Patients Associates to STAT1 Phosphorylation in Monocytes. Front. Immunol. 2022, 13, 932240. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Bischoff, R. Physiology and Pathophysiology of Matrix Metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Mimura, T. Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 905. [Google Scholar] [CrossRef]
- Madkour, M.M.; Anbar, H.S.; El-Gamal, M.I. Current Status and Future Prospects of P38α/MAPK14 Kinase and Its Inhibitors. Eur. J. Med. Chem. 2021, 213, 113216. [Google Scholar] [CrossRef]
- Korb, A.; Tohidast-Akrad, M.; Cetin, E.; Axmann, R.; Smolen, J.; Schett, G. Differential Tissue Expression and Activation of P38 MAPK Alpha, Beta, Gamma, and Delta Isoforms in Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 2745–2756. [Google Scholar] [CrossRef]
- Phull, A.-R.; Nasir, B.; ul Haq, I.; Kim, S.J. Oxidative Stress, Consequences and ROS Mediated Cellular Signaling in Rheumatoid Arthritis. Chem. Biol. Interact. 2018, 281, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Totten, S.P.; Im, Y.K.; Cepeda Cañedo, E.; Najyb, O.; Nguyen, A.; Hébert, S.; Ahn, R.; Lewis, K.; Lebeau, B.; La Selva, R.; et al. STAT1 Potentiates Oxidative Stress Revealing a Targetable Vulnerability That Increases Phenformin Efficacy in Breast Cancer. Nat. Commun. 2021, 12, 3299. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhu, H.; Sheng, F.; Tian, Y.; Zhou, J.; Chen, Y.; Li, S.; Lin, J. Activation of the MAPK11/12/13/14 (P38 MAPK) Pathway Regulates the Transcription of Autophagy Genes in Response to Oxidative Stress Induced by a Novel Copper Complex in HeLa Cells. Autophagy 2014, 10, 1285–1300. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kume, S.; Araki, H.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, S.; Nakagawa, F.; Koya, D.; Haneda, M.; Maegawa, H.; et al. 1-Methylnicotinamide Ameliorates Lipotoxicity-Induced Oxidative Stress and Cell Death in Kidney Proximal Tubular Cells. Free Radic. Biol. Med. 2015, 89, 831–841. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, S.; Chakraborty, D.; Agnihotri, P.; Kumar, V.; Biswas, S. Unveiling the Nexus: Cellular Metabolomics Unravels the Impact of Estrogen on Nicotinamide Metabolism in Mitigating Rheumatoid Arthritis Pathogenesis. Metabolites 2024, 14, 214. https://doi.org/10.3390/metabo14040214
Malik S, Chakraborty D, Agnihotri P, Kumar V, Biswas S. Unveiling the Nexus: Cellular Metabolomics Unravels the Impact of Estrogen on Nicotinamide Metabolism in Mitigating Rheumatoid Arthritis Pathogenesis. Metabolites. 2024; 14(4):214. https://doi.org/10.3390/metabo14040214
Chicago/Turabian StyleMalik, Swati, Debolina Chakraborty, Prachi Agnihotri, Vijay Kumar, and Sagarika Biswas. 2024. "Unveiling the Nexus: Cellular Metabolomics Unravels the Impact of Estrogen on Nicotinamide Metabolism in Mitigating Rheumatoid Arthritis Pathogenesis" Metabolites 14, no. 4: 214. https://doi.org/10.3390/metabo14040214
APA StyleMalik, S., Chakraborty, D., Agnihotri, P., Kumar, V., & Biswas, S. (2024). Unveiling the Nexus: Cellular Metabolomics Unravels the Impact of Estrogen on Nicotinamide Metabolism in Mitigating Rheumatoid Arthritis Pathogenesis. Metabolites, 14(4), 214. https://doi.org/10.3390/metabo14040214