
Integrating Genome Sequencing and Untargeted Metabolomics in Monozygotic Twins with a Rare Complex Neurological Disorder

 , , ,
, , , 
Abstract
:1. Introduction
2. Case Report
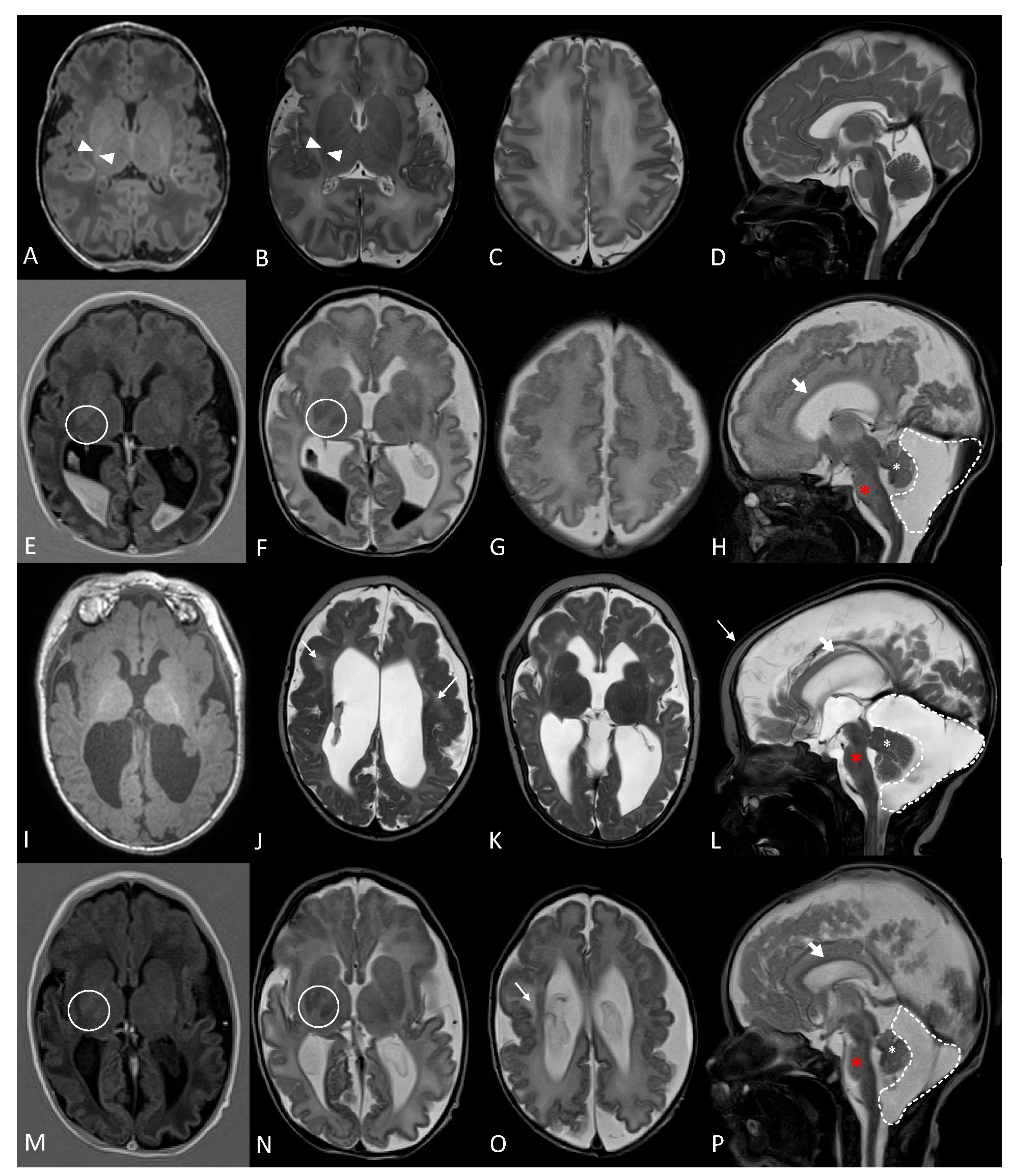
2.1. Clinical Description
2.2. Whole Genome Sequencing Data Analysis
2.3. Untargeted Metabolomics Profile
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruckner-Tuderman, L. Epidemiology of rare diseases is important. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 783–784. [Google Scholar] [CrossRef]
- Ningrum, D.N.A.; Kung, W.-M. Challenges and Perspectives of Neurological Disorders. Brain Sci. 2023, 13, 676. [Google Scholar] [CrossRef]
- Gillard, P.J.; Ganapathy, V.; Graham, G.D.; DiBonaventura, M.D.; Goren, A.; Zorowitz, R.D. Caregiver burden, productivity loss, and indirect costs associated with caring for patients with poststroke spasticity. Clin. Interv. Aging 2015, 10, 1793–1802. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.D.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Erratum to: Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2016, 39, 757. [Google Scholar] [CrossRef]
- Ford, L.; Kennedy, A.D.; Goodman, K.D.; Pappan, K.L.; Evans, A.M.; Miller, L.A.D.; E Wulff, J.; Wiggs, B.R.; Lennon, J.J.; Elsea, S.; et al. Precision of a Clinical Metabolomics Profiling Platform for Use in the Identification of Inborn Errors of Metabolism. J. Appl. Lab. Med. 2020, 5, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Lin, H.-H. The role of GPR56/ADGRG1 in health and disease. Biomed. J. 2021, 44, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Jeong, S.-J.; Yang, A.; Wen, M.; Saslowsky, D.E.; Lencer, W.I.; Araç, D.; Piao, X. Mechanism for adhesion G protein-coupled receptor GPR56-mediated RhoA activation induced by collagen III stimulation. PLoS ONE 2014, 9, e100043. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.; Chang, B.S.; Bodell, A.; Woods, K.; BenZeev, B.; Topcu, M.; Guerrini, R.; Goldberg-Stern, H.; Sztriha, L.; Dobyns, W.B.; et al. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann. Neurol. 2005, 58, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.; Hill, R.S.; Bodell, A.; Chang, B.S.; Basel-Vanagaite, L.; Straussberg, R.; Dobyns, W.B.; Qasrawi, B.; Winter, R.M.; Innes, A.M.; et al. G protein-coupled receptor-dependent development of human frontal cortex. Science 2004, 303, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Adamsky, K.; Vainshtein, A.; Frechter, S.; Dupree, J.L.; Rosenbluth, J.; Peles, E. Caspr and caspr2 are required for both radial and longitudinal organization of myelinated axons. J. Neurosci. 2014, 34, 14820–14826. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Magee, A.; Mahanjah, M.; Vallat, J.-M.; Ouvrier, R.; Abu-Rashid, M.; Mahamid, J.; Schüle, R.; Schulze, M.; Krägeloh-Mann, I.; et al. CNTNAP1 mutations cause CNS hypomyelination and neuropathy with or without arthrogryposis. Neurol. Genet. 2017, 3, e144. [Google Scholar] [CrossRef]
- Garel, P.; Lesca, G.; Ville, D.; Poulat, A.-L.; Chatron, N.; Sanlaville, D.; Portes, V.D.; Arzimanoglou, A.; Lion-François, L. CNTNAP1-encephalopathy: Six novel patients surviving the neonatal period. Eur. J. Paediatr. Neurol. 2022, 37, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Nizon, M.; Cogne, B.; Vallat, J.-M.; Joubert, M.; Liet, J.-M.; Simon, L.; Vincent, M.; Küry, S.; Boisseau, P.; Schmitt, S.; et al. Two novel variants in CNTNAP1 in two siblings presenting with congenital hypotonia and hypomyelinating neuropathy. Eur. J. Hum. Genet. 2017, 25, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Küspert, M.; Bale, T.; Brownstein, C.A.; Towne, M.C.; De Girolami, U.; Shi, J.; Beggs, A.H.; Darras, B.T.; Wegner, M.; et al. Novel mutation in CNTNAP1 results in congenital hypomyelinating neuropathy. Muscle Nerve 2017, 55, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Low, K.; Stals, K.; Caswell, R.; Wakeling, M.; Clayton-Smith, J.; Donaldson, A.; Foulds, N.; Norman, A.; Splitt, M.; Urankar, K.; et al. Phenotype of CNTNAP1: A study of patients demonstrating a specific severe congenital hypomyelinating neuropathy with survival beyond infancy. Eur. J. Hum. Genet. 2018, 26, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Umair, M.; Abbas, S.; Ali, U.; Zaman, G.; Ansar, M.; Wang, R.; Zhang, X.; Houlden, H.; Harlalka, G.V.; et al. Overlapping neurological phenotypes in two extended consanguineous families with novel variants in the CNTNAP1 and ADGRG1 genes. J. Gene Med. 2023, 25, e3513. [Google Scholar] [CrossRef]
- Vallat, J.-M.; Nizon, M.; Magee, A.; Isidor, B.; Magy, L.; Péréon, Y.; Richard, L.; Ouvrier, R.; Cogné, B.; Devaux, J.; et al. Contactin-Associated Protein 1 (CNTNAP1) Mutations Induce Characteristic Lesions of the Paranodal Region. J. Neuropathol. Exp. Neurol. 2016, 75, 1155–1159. [Google Scholar] [CrossRef]
- Kuo, C.; Tsai, M.; Lin, H.; Wang, Y.; Singh, A.K.; Chang, C.; Lin, J.; Hung, P.; Lin, K. Identification and clinical characteristics of a novel missense ADGRG1 variant in bilateral Frontoparietal Polymicrogyria: The electroclinical change from infancy to adulthood after Callosotomy in three siblings. Epilepsia Open 2023, 8, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Sawal, H.A.; Harripaul, R.; Mikhailov, A.; Vleuten, K.; Naeem, F.; Nasr, T.; Hassan, M.J.; Vincent, J.B.; Ayub, M.; Rafiq, M.A. Three Mutations in the Bilateral Frontoparietal Polymicrogyria Gene GPR56 in Pakistani Intellectual Disability Families. J. Pediatr. Genet. 2018, 7, 060–066. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Poirier, K.; Boddaert, N.; Fallet-Bianco, C.; Specchio, N.; Bertini, E.; Caglayan, O.; Lascelles, K.; Elie, C.; Rambaud, J.; et al. GPR56-related bilateral frontoparietal polymicrogyria: Further evidence for an overlap with the cobblestone complex. Brain 2010, 133, 3194–3209. [Google Scholar] [CrossRef]
- Machado, M.C.; Pinheiro da Silva, F. Hyperammonemia due to urea cycle disorders: A potentially fatal condition in the intensive care setting. J. Intensive Care 2014, 2, 22. [Google Scholar] [CrossRef]
- Baba, S.P.; Bhatnagar, A. Role of Thiols in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef]
- Campbell, K.; Vowinckel, J.; Keller, M.A.; Ralser, M. Methionine Metabolism Alters Oxidative Stress Resistance via the Pentose Phosphate Pathway. Antioxid. Redox Signal. 2016, 24, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef]
- Venkataraman, S.S.; Regone, R.; Ammar, H.M.; Govindu, R.R. Pyroglutamic Acidemia: An Underrecognized and Underdiagnosed Cause of High Anion Gap Metabolic Acidosis—A Case Report and Review of Literature. Cureus 2019, 11, e5229. [Google Scholar] [CrossRef] [PubMed]
- Hojjati, M.R.; Li, Z.; Jiang, X.-C. Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim. Biophys. Acta 2005, 1737, 44–51. [Google Scholar] [CrossRef]
- Wang, G.; Krishnamurthy, K.; Bieberich, E. Regulation of primary cilia formation by ceramide. J. Lipid Res. 2009, 50, 2103–2110. [Google Scholar] [CrossRef]
- Wu, D.; Huang, J.; Zhu, H.; Chen, Z.; Chai, Y.; Ke, J.; Lei, K.; Peng, Z.; Zhang, R.; Li, X.; et al. Ciliogenesis requires sphingolipid-dependent membrane and axoneme interaction. Proc. Natl. Acad. Sci. USA 2022, 119, e2201096119. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Altered lipid metabolism in brain injury and disorders. Subcell Biochem. 2008, 49, 241–268. [Google Scholar]
- Li, T.; Chiou, B.; Gilman, C.K.; Luo, R.; Koshi, T.; Yu, D.; Oak, H.C.; Giera, S.; Johnson-Venkatesh, E.; Muthukumar, A.K.; et al. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 2020, 39, e104136. [Google Scholar] [CrossRef]
- Knuplez, E.; Marsche, G. An Updated Review of Pro- and Anti-Inflammatory Properties of Plasma Lysophosphatidylcholines in the Vascular System. Int. J. Mol. Sci. 2020, 21, 4501. [Google Scholar] [CrossRef]
- Giera, S.; Luo, R.; Ying, Y.; Ackerman, S.D.; Jeong, S.J.; Stoveken, H.M.; Folts, C.J.; Welsh, C.A.; Tall, G.G.; Stevens, B.; et al. Microglial transglutaminase-2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. Elife 2018, 7, e33385. [Google Scholar] [CrossRef]
- Chakrapani, A.; Gissen, P.; McKiernan, P. Disorders of Tyrosine Metabolism. In Inborn Metabolic Diseases; Springer: Berlin/Heidelberg, Germany, 2012; pp. 265–276. [Google Scholar] [CrossRef]
- Xue, C.; Li, G.; Zheng, Q.; Gu, X.; Shi, Q.; Su, Y.; Chu, Q.; Yuan, X.; Bao, Z.; Lu, J.; et al. Tryptophan metabolism in health and disease. Cell. Metab. 2023, 35, 1304–1326. [Google Scholar] [CrossRef]
- Kelley, R.E.; Andersson, H.C. Disorders of purines and pyrimidines. Handb. Clin. Neurol. 2014, 120, 827–838. [Google Scholar]
- van Kuilenburg, A.B.; Meinsma, R.; Beke, E.; Assmann, B.; Ribes, A.; Lorente, I.; Busch, R.; Mayatepek, E.; Abeling, N.G.; van Cruchten, A.; et al. beta-Ureidopropionase deficiency: An inborn error of pyrimidine degradation associated with neurological abnormalities. Hum. Mol. Genet. 2004, 13, 2793–2801. [Google Scholar] [CrossRef]
- Glinton, K.E.; Levy, H.L.; Kennedy, A.D.; Pappan, K.L.; Elsea, S.H. Untargeted metabolomics identifies unique though benign biochemical changes in patients with pathogenic variants in UROC1. Mol. Genet. Metab. Rep. 2019, 18, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ainiwaer, A.; Song, Y.; Qin, L.; Peng, A.; Bao, H.; Qin, H. Perturbed gut microbiome and fecal and serum metabolomes are associated with chronic kidney disease severity. Microbiome 2023, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Yadav, D.K. L-Carnitine and Chronic Kidney Disease: A Comprehensive Review on Nutrition and Health Perspectives. J. Pers. Med. 2023, 13, 298. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Noh, J.S.; Yamabe, N.; Kang, K.S.; Tanaka, T.; Yokozawa, T. Beneficial effect of 7-O-galloyl-D-sedoheptulose on oxidative stress and hepatic and renal changes in type 2 diabetic db/db mice. Eur. J. Pharmacol. 2010, 640, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Swann, J. Influence of the Human Gut Microbiome on the Metabolic Phenotype. In The Handbook of Metabolic Phenotyping; Elsevier: Amsterdam, The Netherlands, 2018; pp. 535–560. [Google Scholar] [CrossRef]
- Gatarek, P.; Kaluzna-Czaplinska, J. Trimethylamine N-oxide (TMAO) in human health. EXCLI J. 2021, 20, 301–319. [Google Scholar] [PubMed]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.L.; Stine, J.G.; Bisanz, J.E.; Okafor, C.D.; Patterson, A.D. Bile acids and the gut microbiota: Metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 2023, 21, 236–247. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Chromosome | HGVS DNA Reference | HGVS Protein Reference | Variant Type | ClinGen ID | Zygosity | OMIM Disease |
|---|---|---|---|---|---|---|---|
| ADGRG1 | 16q21 | NM_001145771.3: c.1693C>T | NP_001139243.1: p.Arg565Trp | missense | CA253611 | homozygous | AR cortical dysplasia, complex, with other brain malformations 14A (MIM: 606854) |
| CNTNAP1 | 17q21.2 | NM_003632.3: c.2729A>T | NP_003623.1: p.Glu910Val | missense | CA399649849 | homozygous | AR Hypomyelinating neuropathy (MIM: 618186) AR lethal congenital contracture syndrome (MIM: 616286) |
| ADGRG1 1 | CNTNAP1 1 | Subject 1 | Subject 2 | |
|---|---|---|---|---|
| Head and neck | ||||
| Facial diplegia | - | + | - | - |
| Micrognathia | - | + | retrognathia | NA |
| Macrocephaly | - | - | + | - |
| Microcephaly | - | + | - | - |
| Low-set ears | - | + | - | + |
| Ophthalmologic abnormalities | + | + | - | - |
| Dysmorphic features | - | + | - | - |
| Respiratory | ||||
| Neonatal respiratory insufficiency | - | + | + | + |
| Gastrointestinal | - | + | + | + |
| Absent swallow | ||||
| Tube feeding | - | + | + | + |
| Kidneys | ||||
| Nephromegaly | - | - | + | + (polycystic) |
| Heart | ||||
| Cardiac arrythmias | - | - | + | + |
| ASD | - | - | + | + |
| Skeletal | ||||
| Contractures | - | some | - | - |
| Clenched hands | - | + | - | - |
| Foot deformities | - | + | - | - |
| Polydactyly | - | - | + | + |
| Muscle | ||||
| Hypotonia | + | + | + | + |
| Increased muscle tone/spasticity | + | + | + | + |
| Neurologic | ||||
| DD/ID | + | + | + | + |
| Psychomotor delay | + | + | + | + |
| Hyperreflexia (CNS) | + | + | + (clonus) | + (clonus) |
| Hypomyelination | Dysmyelination | + | + | + |
| Seizures | + | some | + | + |
| Abnormal basal ganglia | - | + | + | + |
| Brainstem and cerebellar hypoplasia | + | + | + | NA |
| Reduced cerebral volume | - | + | + | + |
| Thin corpus callosum | + | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaath, R.; Al-Maraghi, A.; Ali, H.; AlRayahi, J.; Kennedy, A.D.; DeBalsi, K.L.; Hussein, S.; Elbashir, N.; Padmajeya, S.S.; Palaniswamy, S.; et al. Integrating Genome Sequencing and Untargeted Metabolomics in Monozygotic Twins with a Rare Complex Neurological Disorder. Metabolites 2024, 14, 152. https://doi.org/10.3390/metabo14030152
Shaath R, Al-Maraghi A, Ali H, AlRayahi J, Kennedy AD, DeBalsi KL, Hussein S, Elbashir N, Padmajeya SS, Palaniswamy S, et al. Integrating Genome Sequencing and Untargeted Metabolomics in Monozygotic Twins with a Rare Complex Neurological Disorder. Metabolites. 2024; 14(3):152. https://doi.org/10.3390/metabo14030152
Chicago/Turabian StyleShaath, Rulan, Aljazi Al-Maraghi, Haytham Ali, Jehan AlRayahi, Adam D. Kennedy, Karen L. DeBalsi, Sura Hussein, Najwa Elbashir, Sujitha S. Padmajeya, Sasirekha Palaniswamy, and et al. 2024. "Integrating Genome Sequencing and Untargeted Metabolomics in Monozygotic Twins with a Rare Complex Neurological Disorder" Metabolites 14, no. 3: 152. https://doi.org/10.3390/metabo14030152
APA StyleShaath, R., Al-Maraghi, A., Ali, H., AlRayahi, J., Kennedy, A. D., DeBalsi, K. L., Hussein, S., Elbashir, N., Padmajeya, S. S., Palaniswamy, S., Elsea, S. H., Akil, A. A., Yousri, N. A., & Fakhro, K. A. (2024). Integrating Genome Sequencing and Untargeted Metabolomics in Monozygotic Twins with a Rare Complex Neurological Disorder. Metabolites, 14(3), 152. https://doi.org/10.3390/metabo14030152