
Palmitate Compromises C6 Astrocytic Cell Viability and Mitochondrial Function

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Fatty Acid Conjugation with BSA
2.4. Assessment of Cell Viability
2.5. Assessment of Cellular Morphology
2.6. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.7. Mitochondrial Membrane Potential
2.8. Measurement of Mitochondrial Respiration
2.9. Measurement of Mitochondrial Respiratory Complex Activity
2.10. ROS Generation
2.11. TBARS—Thiobarbituric Acid Reactive Species Levels
2.12. Statistical Analysis
3. Results
3.1. Effect of Saturated Fatty Acids on Cellular Viability and Morphology in Astrocytes
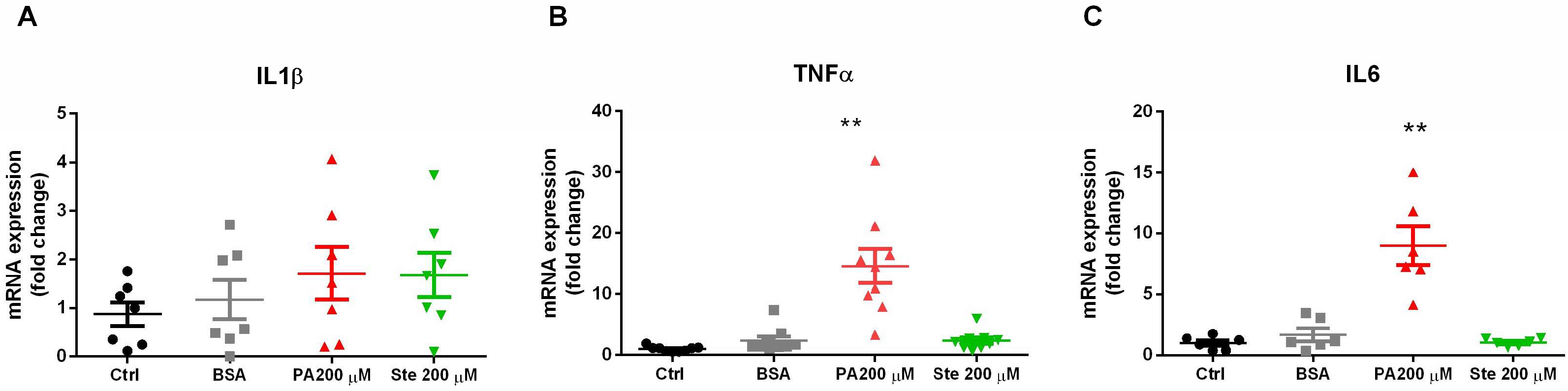
3.2. Palmitate Increases Gene Expression of Pro-Inflammatory Cytokines in Astrocytes
3.3. Palmitate Reduces Mitochondrial Membrane Potential in Astrocytes
3.4. Palmitate Decreases the Expression of Mfn2 and Citrate Synthase (CS)
3.5. Palmitate Decreases the Activity of Mitochondrial Complex I
3.6. Palmitate Does Not Change the Mitochondrial Respiration in Astrocytes
3.7. Palmitate Does Not Induce Oxidative Stress in Astrocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| ATP | Adenosine triphosphate |
| BBB | Blood–brain barrier |
| BMI | Body mass index |
| BSA | Bovine serum albumin |
| CNS | Central nervous system |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DCF | Dichlorofluorescein |
| DCFH | Dichlorodihydrofluorescein |
| DCFH-DA | 2,7-dichlorodihydrofluorescein diacetate |
| DCIP | 2,6-dicloindophenol |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | Dimethylsulfoxide |
| FBS | Fetal bovine serum |
| FFA | Free fatty acids |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GFAP | Glial fibrillary acid protein |
| GLUT1 | Glucose transporter 1 |
| HFD | High-fat diet |
| IL-1β | Interleukin 1 β |
| IL-6 | Interleukin 6 |
| LC3B-II | Microtubule-associated protein 1A/1B-light chain 3 conjugated with phosphatidylethanolamine |
| Mfn2 | Mitofusin-2 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium |
| NADH | Nicotinamide adenine dinucleotide |
| PA | Palmitate |
| PBS | Phosphate-buffered saline |
| ROX | Extramitochondrial respiration |
| SEM | Standard error of the mean |
| Ste | Stearate |
| TBARS | Thiobarbinturic-acid-reactive substances |
| TNF-α | Tumor necrosis factor-α |
| UCP-2 | Uncoupler protein-2 |
| ROS | Reactive oxygen species |
References
- WHO. WHO Guidelines on Drawing Blood: Best Practices in Phlebotomy; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef]
- Gaspar, J.M.; Baptista, F.I.; Macedo, M.P.; Ambrósio, A.F. Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. ACS Chem. Neurosci. 2016, 7, 131–142. [Google Scholar] [CrossRef]
- Boccara, E.; Golan, S.; Beeri, M.S. The association between regional adiposity, cognitive function, and dementia-related brain changes: A systematic review. Front. Med. 2023, 10, 1160426. [Google Scholar] [CrossRef]
- Cope, E.C.; LaMarca, E.A.; Monari, P.K.; Olson, L.B.; Martinez, S.; Zych, A.D.; Katchur, N.J.; Gould, E. Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J. Neurosci. 2018, 38, 8889–8904. [Google Scholar] [CrossRef]
- Atlantis, E.; Baker, M. Obesity effects on depression: Systematic review of epidemiological studies. Int. J. Obes. 2008, 32, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Musselman, D.L.; Betan, E.; Larsen, H.; Phillips, L.S. Relationship of depression to diabetes types 1 and 2: Epidemiology, biology, and treatment. Biol. Psychiatry 2003, 54, 317–329. [Google Scholar] [CrossRef]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.; Dias, C.; Lourenço, C.F.; Laranjinha, J.; de Bem, A.; Ledo, A. A High Fat/Cholesterol Diet Recapitulates Some Alzheimer’s Disease-Like Features in Mice: Focus on Hippocampal Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2021, 82, 1619–1633. [Google Scholar] [CrossRef]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 137309. [Google Scholar] [CrossRef]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Melo, H.M.; Silva, G.d.S.S.d.; Sant’ana, M.R.; Teixeira, C.V.L.; Clarke, J.R.; Coreixas, V.S.M.; de Melo, B.C.; Fortuna, J.T.; Forny-Germano, L.; Ledo, J.H.; et al. Palmitate Is Increased in the Cerebrospinal Fluid of Humans with Obesity and Induces Memory Impairment in Mice via Pro-inflammatory TNF-α. Cell Rep. 2020, 30, 2180–2194.e8. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Melrose, J.; Chan, C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Sarman, B.; García-Cáceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.d.A.B.; Melo, N.d.F.M.; Vieira, É.D.S.; Nogueira, P.A.S.; Coope, A.; Velloso, L.A.; Dezonne, R.S.; Ueira-Vieira, C.; Botelho, F.V.; Gomes, J.d.A.S.; et al. Palmitate treated-astrocyte conditioned medium contains increased glutathione and interferes in hypothalamic synaptic network in vitro. Neurochem. Int. 2018, 120, 140–148. [Google Scholar] [CrossRef]
- Ortiz-Rodriguez, A.; Acaz-Fonseca, E.; Boya, P.; Arevalo, M.A.; Garcia-Segura, L.M. Lipotoxic Effects of Palmitic Acid on Astrocytes Are Associated with Autophagy Impairment. Mol. Neurobiol. 2019, 56, 1665–1680. [Google Scholar] [CrossRef]
- Ng, Y.-W.; Say, Y.-H. Palmitic acid induces neurotoxicity and gliatoxicity in SH-SY5Y human neuroblastoma and T98G human glioblastoma cells. PeerJ 2018, 6, e4696. [Google Scholar] [CrossRef]
- Alsabeeh, N.; Chausse, B.; Kakimoto, P.A.; Kowaltowski, A.J.; Shirihai, O. Cell culture models of fatty acid overload: Problems and solutions. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2018, 1863, 143–151. [Google Scholar] [CrossRef]
- Cassina, A.; Radi, R. Differential Inhibitory Action of Nitric Oxide and Peroxynitrite on Mitochondrial Electron Transport. Arch. Biochem. Biophys. 1996, 328, 309–316. [Google Scholar] [CrossRef]
- Fischer, J.C.; Ruitenbeek, W.; Berden, J.A.; Trijbels, J.; Veerkamp, J.H.; Stadhouders, A.M.; Sengers, R.C.; Janssen, A.J. Differential investigation of the capacity of succinate oxidation in human skeletal muscle. Clin. Chim. Acta 1985, 153, 23–36. [Google Scholar] [CrossRef]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gérard, B.; Rötig, A.; Saudubray, J.; Munnich, A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Bourre, J.M.; Gozlan-Devillierre, N.; Daudu, O.; Baumann, N. Is there a blood-brain relationship for saturated fatty acids during development? Biol. Neonate 1978, 34, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Mietus, L.J. Palmitate and Cholesterol Transport Through the Blood-Brain Barrier. J. Neurochem. 1980, 34, 463–466. [Google Scholar] [CrossRef]
- Rodriguez-Navas, C.; Morselli, E.; Clegg, D.J. Sexually dimorphic brain fatty acid composition in low and high fat diet-fed mice. Mol. Metab. 2016, 5, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- González-Giraldo, Y.; Garcia-Segura, L.M.; Echeverria, V.; Barreto, G.E. Tibolone Preserves Mitochondrial Functionality and Cell Morphology in Astrocytic Cells Treated with Palmitic Acid. Mol. Neurobiol. 2018, 55, 4453–4462. [Google Scholar] [CrossRef]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Wang, J.; Liu, S.; Gao, M.; Ling, E.; Hao, A. Cytoprotective effects of melatonin on astroglial cells subjected to palmitic acid treatment in vitro. J. Pineal Res. 2012, 52, 253–264. [Google Scholar] [CrossRef]
- de Andrade, A.M.; Fernandes, M.d.C.; de Fraga, L.S.; Porawski, M.; Giovenardi, M.; Guedes, R.P. Omega-3 fatty acids revert high-fat diet-induced neuroinflammation but not recognition memory impairment in rats. Metab. Brain Dis. 2017, 32, 1871–1881. [Google Scholar] [CrossRef]
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 285. [Google Scholar] [CrossRef]
- Galland, F.; Seady, M.; Taday, J.; Smaili, S.S.; Gonçalves, C.A.; Leite, M.C. Astrocyte culture models: Molecular and function characterization of primary culture, immortalized astrocytes and C6 glioma cells. Neurochem. Int. 2019, 131, 104538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Seitz, L.C.; Abramczyk, A.M.; Chan, C. Synergistic effect of cAMP and palmitate in promoting altered mitochondrial function and cell death in HepG2 cells. Exp. Cell Res. 2010, 316, 716–727. [Google Scholar] [CrossRef]
- Martin-Jiménez, C.; González, J.; Vesga, D.; Aristizabal, A.; Barreto, G.E. Tibolone Ameliorates the Lipotoxic Effect of Palmitic Acid in Normal Human Astrocytes. Neurotox. Res. 2020, 38, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Nie, S.-D.; Qu, M.-L.; Zhou, D.; Wu, L.-Y.; Shi, X.-J.; Ma, L.-R.; Li, X.; Zhou, S.-L.; Wang, S.; et al. The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 2018, 9, 771. [Google Scholar] [CrossRef]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Galizzi, G.; Amato, A.; Terzo, S.; Picone, P.; Cristaldi, L.; Mulè, F.; Di Carlo, M. Regular Intake of Pistachio Mitigates the Deleterious Effects of a High Fat-Diet in the Brain of Obese Mice. Antioxidants 2020, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Corem, N.; Anzi, S.; Gelb, S.; Ben-Zvi, A. Leptin receptor deficiency induces early, transient and hyperglycaemia-independent blood-brain barrier dysfunction. Sci. Rep. 2019, 9, 2884. [Google Scholar] [CrossRef]
- Berridge, M.; Tan, A. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Wojtczak, L.; Schönfeld, P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim. Biophys. Acta (BBA)-Bioenerg. 1993, 1183, 41–57. [Google Scholar] [CrossRef]
- Broekemeier, K.M.; Pfeiffer, D.R. Inhibition of The Mitochondrial Permeability Transition by Cyclosporin A during Long Time Frame Experiments: Relationship between Pore Opening and the Activity of Mitochondrial Phospholipases. Biochemistry 1995, 34, 16440–16449. [Google Scholar] [CrossRef] [PubMed]
- Więckowski, M.R.; Wojtczak, L. Fatty acid-induced uncoupling of oxidative phosphorylation is partly due to opening of the mitochondrial permeability transition pore. FEBS Lett. 1998, 423, 339–342. [Google Scholar] [CrossRef]
- Andreyev, A.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Volkov, N.I. Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS Lett. 1988, 226, 265–269. [Google Scholar] [CrossRef] [PubMed]
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood–Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.-M.; Xu, A.-M.; Dai, R.-P.; Zhou, Z.-G. Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef]
- Hansen, C.; Olsen, K.; Pilegaard, H.; Bangsbo, J.; Gliemann, L.; Hellsten, Y. High metabolic substrate load induces mitochondrial dysfunction in rat skeletal muscle microvascular endothelial cells. Physiol. Rep. 2021, 9, e14855. [Google Scholar] [CrossRef]
- Maneechote, C.; Chunchai, T.; Apaijai, N.; Chattipakorn, N.; Chattipakorn, S.C. Pharmacological Targeting of Mitochondrial Fission and Fusion Alleviates Cognitive Impairment and Brain Pathologies in Pre-diabetic Rats. Mol. Neurobiol. 2022, 59, 3690–3702. [Google Scholar] [CrossRef]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell. Endocrinol. 2018, 460, 238–245. [Google Scholar] [CrossRef]
- Yang, C.; Sui, G.; Li, D.; Wang, L.; Zhang, S.; Lei, P.; Chen, Z.; Wang, F. Exogenous IGF-1 alleviates depression-like behavior and hippocampal mitochondrial dysfunction in high-fat diet mice. Physiol. Behav. 2021, 229, 113236. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene ID |
|---|---|
| Gapdh | 14433 transcript NM_001411840.1 |
| Il1b | 16176 transcript NM_008361.4 |
| Il6 | 16193 transcript NM_031168.2 |
| Tnf | 21926 transcript NM_013693.3 |
| Mfn2 | 170731 transcript NM_133201.3 |
| CS | 12974 transcript NM_026444.4 |
| Acaca | 107476 transcript NM_133360.3 |
| Opa1 | 74143 transcript NM_133752.4 |
| Becn1 | 56208 transcript NM_019584.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitt, L.O.; Blanco, A.; Lima, S.V.; Mancini, G.; Mendes, N.F.; Latini, A.; Gaspar, J.M. Palmitate Compromises C6 Astrocytic Cell Viability and Mitochondrial Function. Metabolites 2024, 14, 161. https://doi.org/10.3390/metabo14030161
Schmitt LO, Blanco A, Lima SV, Mancini G, Mendes NF, Latini A, Gaspar JM. Palmitate Compromises C6 Astrocytic Cell Viability and Mitochondrial Function. Metabolites. 2024; 14(3):161. https://doi.org/10.3390/metabo14030161
Chicago/Turabian StyleSchmitt, Luisa O., Antonella Blanco, Sheila V. Lima, Gianni Mancini, Natalia F. Mendes, Alexandra Latini, and Joana M. Gaspar. 2024. "Palmitate Compromises C6 Astrocytic Cell Viability and Mitochondrial Function" Metabolites 14, no. 3: 161. https://doi.org/10.3390/metabo14030161
APA StyleSchmitt, L. O., Blanco, A., Lima, S. V., Mancini, G., Mendes, N. F., Latini, A., & Gaspar, J. M. (2024). Palmitate Compromises C6 Astrocytic Cell Viability and Mitochondrial Function. Metabolites, 14(3), 161. https://doi.org/10.3390/metabo14030161