Metabolic Profiling of Retrograde Pathway Transcription Factors Rtg1 and Rtg3 Knockout Yeast


Abstract
:1. Introduction
2. Results and Discussion
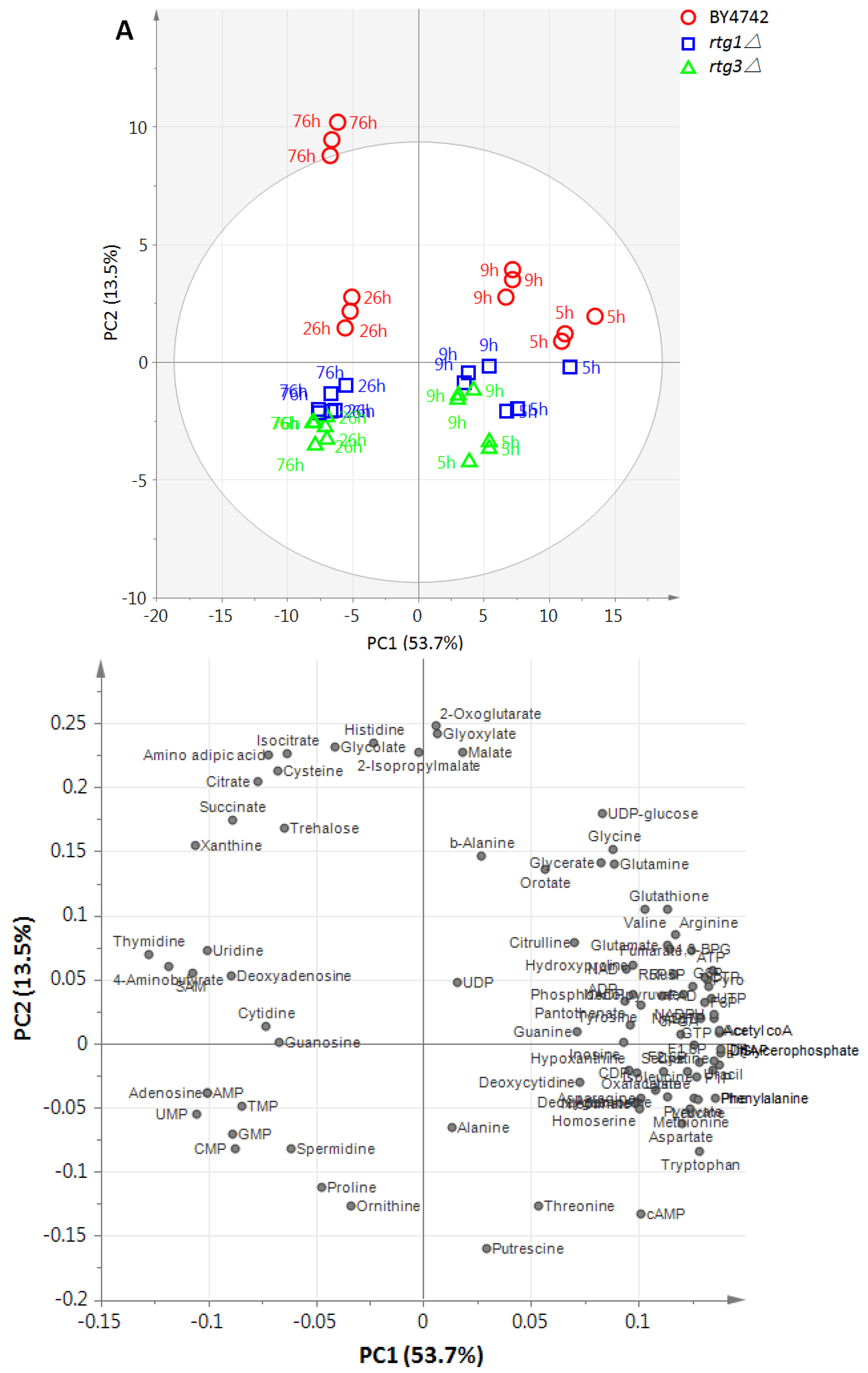
2.1. Time-Course Metabolic Profiling of RTG-Deleted Strains

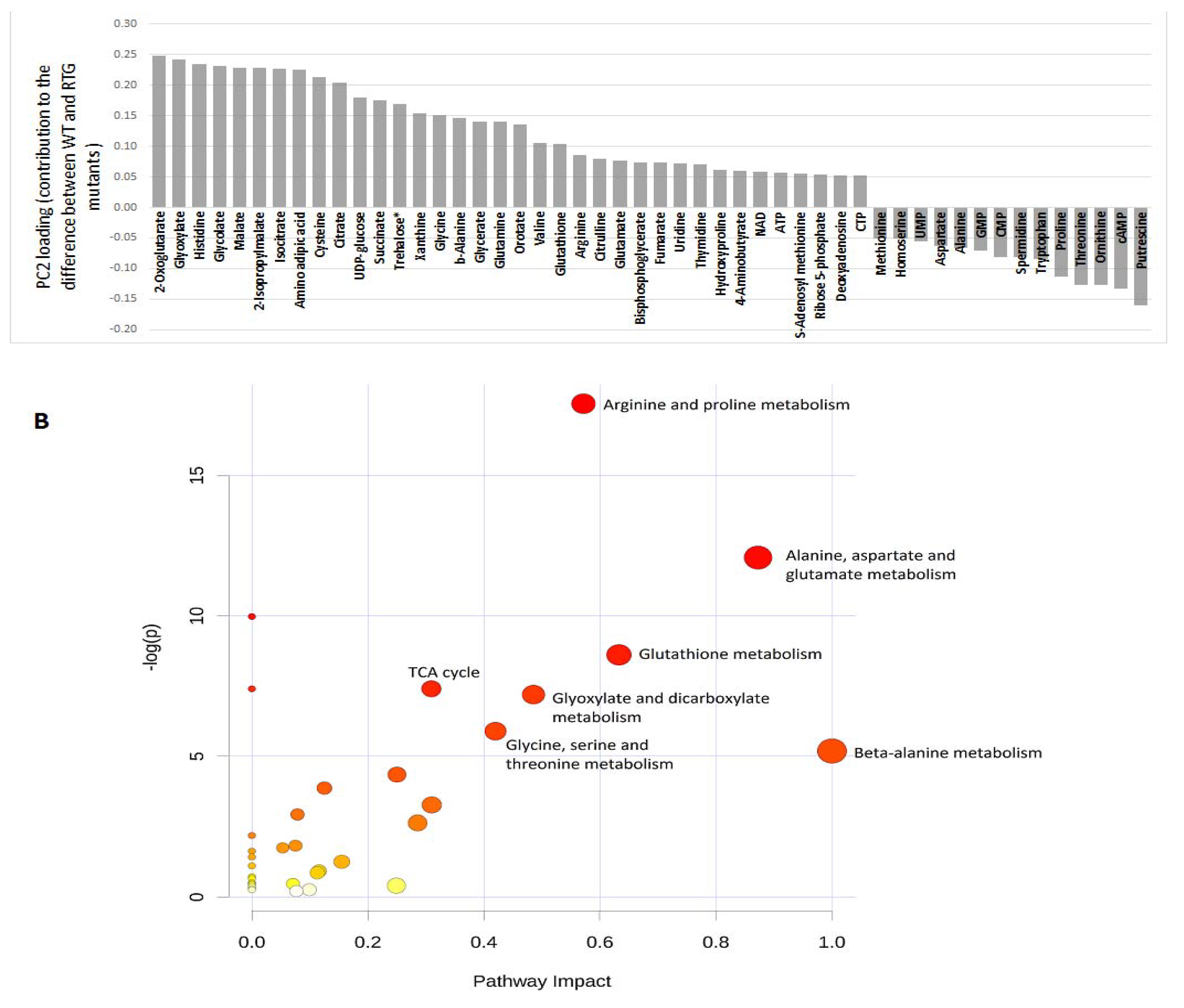
2.2. Metabolites and Metabolic Pathways Associated with RTG1 and RTG3



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | 5 h | 9 h | 26 h | 76 h | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rtg1∆ | rtg3∆ | p | rtg1∆ | rtg3∆ | p | rtg1∆ | rtg3∆ | p | rtg1∆ | rtg3∆ | p | |||||||||||||
| TCA/glyoxylate cycle | ||||||||||||||||||||||||
| 2-Oxoglutarate | −9.26 | −15.92 | 0.179 | −8.32 | −9.48 | 0.682 | −3.79 | −5.07 | 0.205 | −4.54 | −6.28 | 0.356 | ||||||||||||
| Malate | −1.23 | −1.51 | 0.308 | −1.14 | −1.01 | 0.647 | −1.69 | −1.76 | 0.434 | −2.45 | −2.68 | 0.273 | ||||||||||||
| Isocitrate | −3.47 | −3.89 | 0.679 | −1.17 | −1.55 | 0.721 | −5.15 | −7.50 | 0.200 | −5.94 | −10.86 | 0.264 | ||||||||||||
| Citrate | −1.69 | −2.73 | 0.363 | −1.37 | −1.51 | 0.686 | −2.11 | −4.42 | 0.206 | −2.35 | −3.31 | 0.306 | ||||||||||||
| Succinate | 1.09 | −1.25 | 0.358 | −1.20 | −1.21 | 0.980 | −1.16 | −1.19 | 0.830 | −1.97 | −2.11 | 0.530 | ||||||||||||
| Fumarate | −1.23 | −1.35 | 0.377 | −1.10 | −1.16 | 0.699 | 1.29 | 1.12 | 0.351 | −1.34 | −1.94 | 0.304 | ||||||||||||
| Glyoxylate | 1.09 | −1.39 | 0.352 | −1.21 | −1.26 | 0.620 | −1.77 | −1.75 | 0.981 | −6.40 | −6.00 | 0.792 | ||||||||||||
| Glycolate | −1.20 | −1.22 | 0.885 | −1.31 | −1.23 | 0.665 | −1.23 | −1.06 | 0.236 | −5.50 | −6.52 | 0.376 | ||||||||||||
| Starch and sucrose metabolism | ||||||||||||||||||||||||
| UDP−glucose | −1.15 | −1.46 | 0.377 | −1.20 | −1.34 | 0.719 | −1.21 | −1.56 | 0.332 | −1.25 | −1.50 | 0.330 | ||||||||||||
| Trehalose | −1.08 | −1.11 | 0.680 | 1.17 | 1.07 | 0.537 | 1.29 | 1.04 | 0.303 | −2.23 | −2.70 | 0.362 | ||||||||||||
| Pyrimidine metabolism | ||||||||||||||||||||||||
| Orotate | −1.40 | −2.21 | 0.341 | −1.25 | −1.39 | 0.668 | 1.05 | 1.01 | 0.622 | −1.08 | −1.15 | 0.733 | ||||||||||||
| Uridine | −1.48 | 1.18 | 0.385 | 1.31 | 1.96 | 0.659 | −1.24 | −1.26 | 0.987 | −1.03 | −1.05 | 0.907 | ||||||||||||
| Thymidine | −1.19 | 1.03 | 0.376 | −1.02 | 1.03 | 0.954 | 1.26 | 1.26 | 0.982 | −1.26 | −1.32 | 0.802 | ||||||||||||
| CTP | −1.27 | −1.55 | 0.363 | −1.34 | −1.36 | 0.953 | −1.84 | −3.12 | 0.214 | −3.51 | −4.69 | 0.341 | ||||||||||||
| UMP | −1.28 | −1.72 | 0.398 | −1.44 | −1.37 | 0.819 | 1.26 | 1.60 | 0.333 | 2.12 | 1.78 | 0.336 | ||||||||||||
| CMP | −1.17 | −1.23 | 0.867 | −1.25 | −1.25 | 0.999 | 1.71 | 1.77 | 0.831 | 2.25 | 2.16 | 0.808 | ||||||||||||
| Purine metabolism | ||||||||||||||||||||||||
| Xanthine | −1.08 | 1.58 | 0.374 | −1.08 | 1.14 | 0.646 | −1.24 | −1.51 | 0.217 | −1.92 | −2.65 | 0.376 | ||||||||||||
| ATP | −1.14 | −1.33 | 0.385 | −1.24 | −1.37 | 0.660 | −1.38 | −2.64 | 0.196 | −2.31 | −3.81 | 0.298 | ||||||||||||
| Deoxyadenosine | −1.57 | −1.77 | 0.589 | −1.04 | 1.10 | 0.675 | 1.24 | 1.36 | 0.325 | −1.28 | −1.17 | 0.434 | ||||||||||||
| GMP | −1.15 | −1.82 | 0.390 | −1.06 | −1.05 | 0.938 | 1.41 | 1.95 | 0.232 | 2.64 | 2.34 | 0.522 | ||||||||||||
| cAMP | −1.02 | 1.03 | 0.864 | 1.20 | 1.29 | 0.787 | 1.01 | 1.63 | 0.287 | 4.27 | 4.43 | 0.800 | ||||||||||||
| Amino acid metabolism | ||||||||||||||||||||||||
| Histidine metabolism | ||||||||||||||||||||||||
| Histidine | −1.18 | −1.34 | 0.339 | −1.30 | −1.50 | 0.586 | −2.52 | −2.46 | 0.851 | −2.62 | −2.24 | 0.264 | ||||||||||||
| Cysteine and methionine metabolism | ||||||||||||||||||||||||
| Cysteine | −1.50 | −2.62 | 0.210 | −1.11 | 1.07 | 0.570 | 1.17 | 1.03 | 0.505 | −3.34 | −3.58 | 0.800 | ||||||||||||
| Methionine | −1.11 | −1.34 | 0.312 | 1.26 | 1.14 | 0.651 | 1.03 | 1.16 | 0.228 | 3.87 | 4.03 | 0.539 | ||||||||||||
| S−Adenosylmethionine | 1.15 | 1.02 | 0.840 | 2.40 | 2.29 | 0.924 | 1.33 | 2.12 | 0.212 | −1.62 | 1.08 | 0.352 | ||||||||||||
| Valine, leucine and isoleucine metabolism | ||||||||||||||||||||||||
| 2−Isopropylmalate | −1.54 | −2.13 | 0.377 | −2.07 | −2.48 | 0.735 | −1.37 | −1.66 | 0.380 | −1.64 | −1.87 | 0.434 | ||||||||||||
| Valine | −1.28 | −1.58 | 0.235 | −1.24 | −1.36 | 0.795 | −1.09 | 1.00 | 0.281 | −1.26 | −1.24 | 0.797 | ||||||||||||
| Lysine metabolism | ||||||||||||||||||||||||
| Amino adipic acid | −5.14 | −11.67 | 0.369 | −12.36 | −14.42 | 0.644 | −2.47 | −3.68 | 0.190 | −5.15 | −7.05 | 0.339 | ||||||||||||
| Glycine, serine and threonine metabolism | ||||||||||||||||||||||||
| Glycine | −1.24 | −1.64 | 0.354 | −1.26 | −1.18 | 0.812 | −1.31 | −1.25 | 0.835 | −1.32 | −1.36 | 0.588 | ||||||||||||
| Glycerate | −1.09 | −1.23 | 0.547 | −1.15 | −1.19 | 0.936 | −1.13 | −1.01 | 0.340 | −1.50 | −1.85 | 0.288 | ||||||||||||
| Homoserine | 1.31 | −1.05 | 0.342 | 1.08 | 1.36 | 0.687 | 1.09 | −1.10 | 0.323 | 1.17 | −1.16 | 0.320 | ||||||||||||
| Threonine | 1.18 | −1.02 | 0.345 | 1.49 | 1.60 | 0.740 | 1.85 | 1.77 | 0.248 | 1.79 | 1.56 | 0.285 | ||||||||||||
| Beta-alanine metabolism | ||||||||||||||||||||||||
| b-Alanine | 1.34 | −1.17 | 0.384 | −1.30 | −2.01 | 0.673 | 1.07 | 1.26 | 0.836 | −2.98 | −2.72 | 0.798 | ||||||||||||
| Arginine and proline metabolism | ||||||||||||||||||||||||
| Arginine | 1.09 | −1.04 | 0.395 | −1.41 | −1.51 | 0.868 | −1.43 | −1.69 | 0.307 | −2.00 | −2.53 | 0.330 | ||||||||||||
| Citrulline | −35.03 | −37.33 | 0.842 | −12.89 | −10.94 | 0.669 | −3.39 | −5.10 | 0.209 | −3.48 | −4.42 | 0.595 | ||||||||||||
| Hydroxyproline | −1.06 | −1.61 | 0.090 | −1.05 | 1.02 | 0.938 | −1.08 | −1.46 | 0.325 | −1.69 | −1.15 | 0.264 | ||||||||||||
| 4-Aminobutyrate | −4.65 | −5.55 | 0.689 | −1.76 | −1.37 | 0.678 | −2.28 | −1.48 | 0.232 | 1.14 | −1.05 | 0.636 | ||||||||||||
| Proline | −1.34 | −1.45 | 0.817 | −1.42 | −1.25 | 0.682 | −1.17 | 1.08 | 0.215 | 2.91 | 3.78 | 0.302 | ||||||||||||
| Alanine, aspartate and glutamate metabolism | ||||||||||||||||||||||||
| Glutamine | −2.01 | −2.53 | 0.306 | −2.25 | −2.62 | 0.718 | −2.02 | −2.61 | 0.328 | −1.93 | −2.84 | 0.340 | ||||||||||||
| Glutamate | −2.04 | −2.76 | 0.369 | −2.27 | −2.63 | 0.189 | −1.57 | −1.93 | 0.237 | −1.94 | −2.19 | 0.266 | ||||||||||||
| Aspartate | 1.27 | 1.13 | 0.387 | 1.13 | 1.16 | 0.678 | 1.01 | −1.20 | 0.179 | 1.41 | 1.16 | 0.286 | ||||||||||||
| Alanine | −1.05 | −1.23 | 0.355 | 1.08 | 1.07 | 0.989 | 1.21 | 1.22 | 0.967 | 1.86 | 1.71 | 0.286 | ||||||||||||
| Phenylalanine, tyrosine and tryptophan metabolism | ||||||||||||||||||||||||
| Tryptophan | −1.11 | −1.26 | 0.336 | −1.08 | −1.14 | 0.714 | 1.38 | 1.56 | 0.218 | 1.79 | 1.97 | 0.275 | ||||||||||||
| Glutathione metabolism, polyamine biosynthesis | ||||||||||||||||||||||||
| Glutathione | −1.21 | −1.43 | 0.344 | −1.21 | −1.27 | 0.640 | −1.03 | −1.07 | 0.272 | −1.36 | −1.51 | 0.332 | ||||||||||||
| Spermidine | −1.33 | −1.74 | 0.674 | −1.12 | −1.37 | 0.675 | 3.01 | 3.56 | 0.641 | 13.24 | 20.61 | 0.383 | ||||||||||||
| Ornithine | −1.87 | 1.13 | 0.002 | 1.45 | 1.81 | 0.110 | −1.15 | 1.09 | 0.251 | 1.90 | 2.21 | 0.334 | ||||||||||||
| Putrescine | 1.83 | 2.32 | 0.343 | 1.75 | 2.13 | 0.723 | 2.28 | 1.68 | 0.324 | 5.33 | 4.85 | 0.732 | ||||||||||||
| Pentose phosphate pathway | ||||||||||||||||||||||||
| Ribose 5-phosphate | −1.41 | −2.46 | 0.341 | −1.41 | −1.58 | 0.616 | −1.98 | −2.06 | 0.934 | −2.67 | −2.47 | 0.651 | ||||||||||||
| Glycolysis | ||||||||||||||||||||||||
| Bisphosphoglycerate | −1.23 | −2.25 | 0.304 | −1.64 | −1.62 | 0.985 | −1.92 | −2.48 | 0.522 | −3.12 | −3.99 | 0.446 | ||||||||||||
| Others (co-factors) | ||||||||||||||||||||||||
| NAD | −1.20 | −1.37 | 0.328 | −1.23 | −1.24 | 0.932 | −1.39 | −1.51 | 0.294 | −1.29 | −1.39 | 0.343 | ||||||||||||
2.3. Metabolic Alteration Levels in RTG1 and RTG3

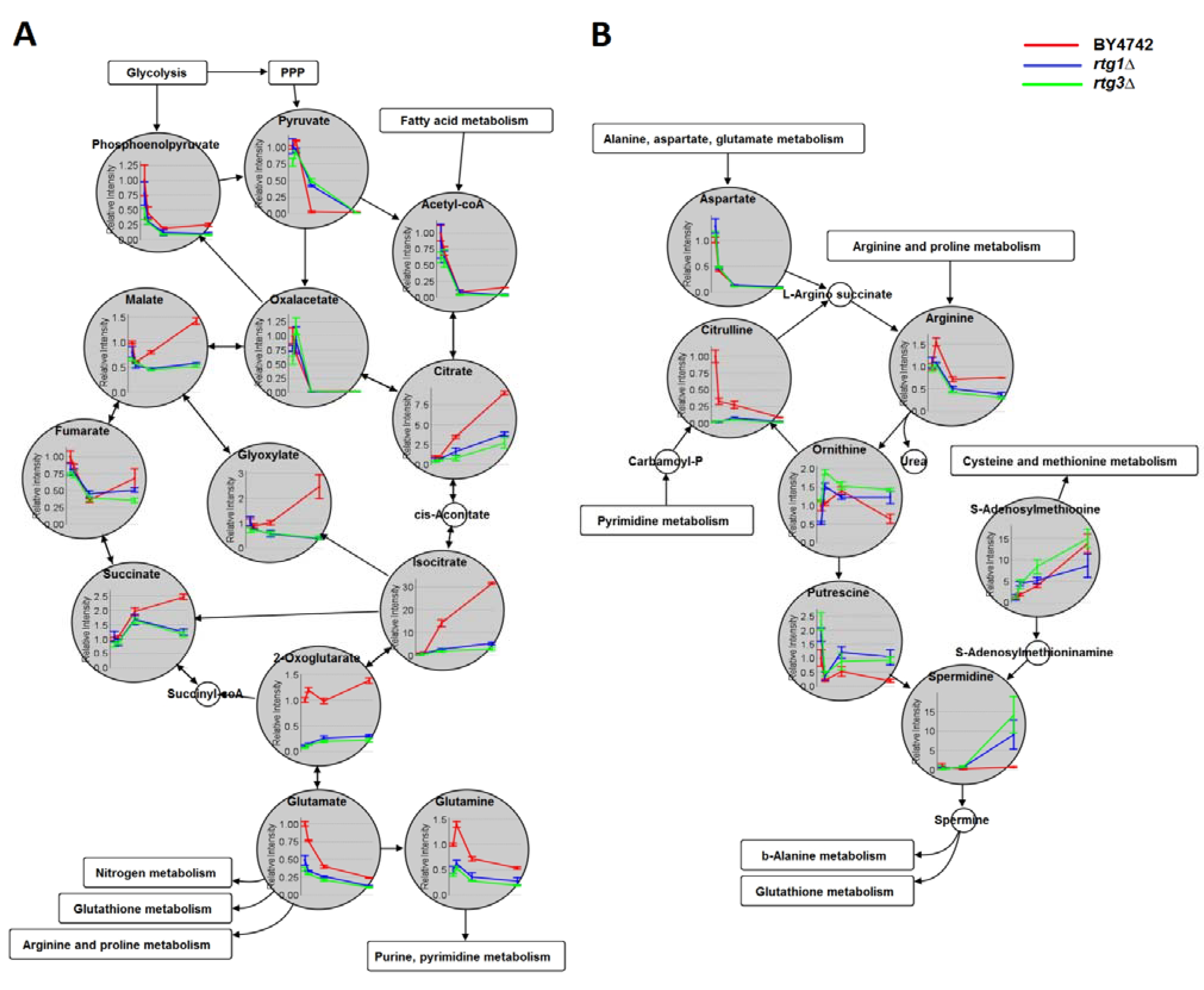
2.4. Comparison with Previous Studies
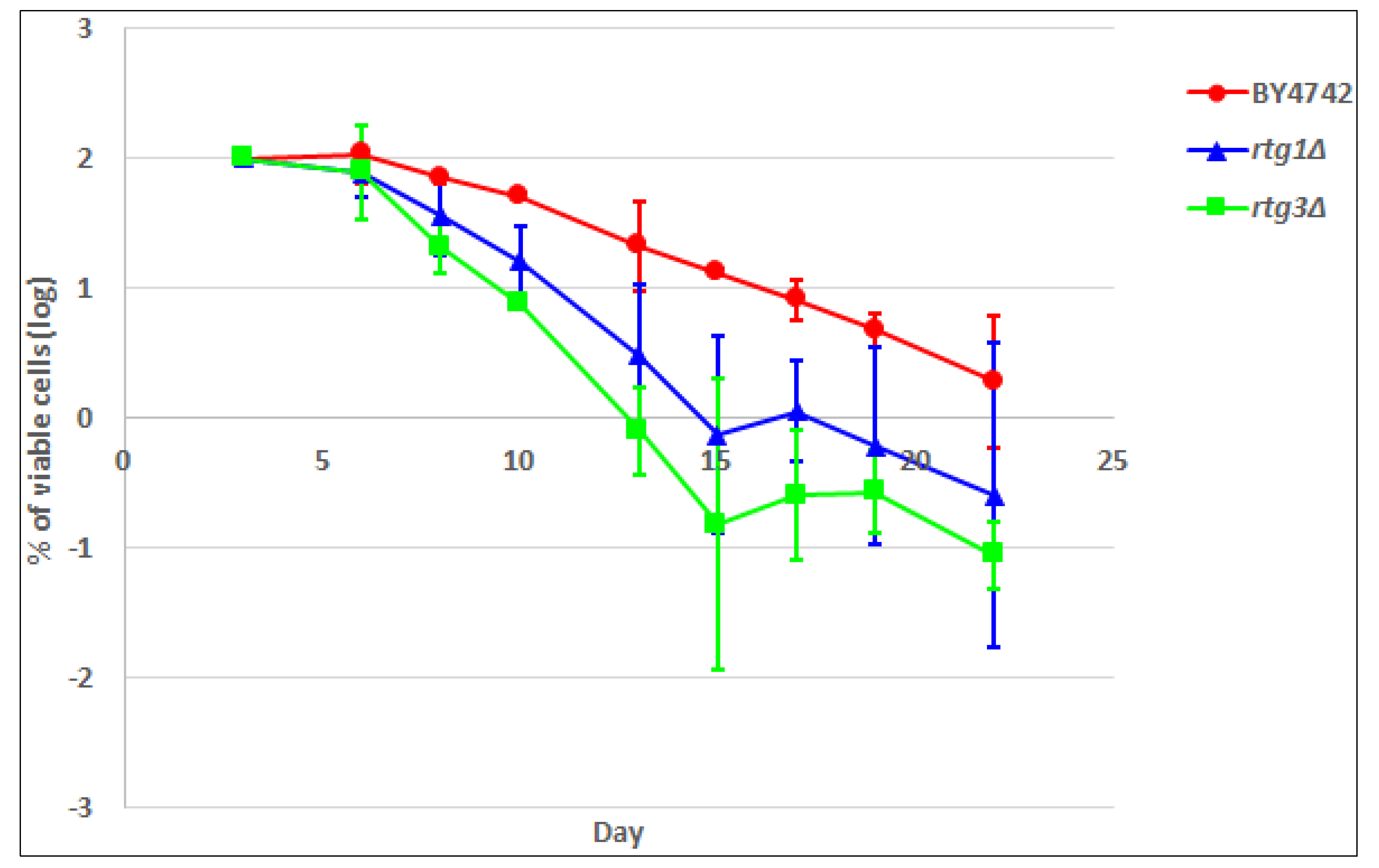
2.5. Yeast Chronological Lifespan and Its Relation with RTG1 and RTG3

3. Experimental Section
3.1. Strain Growth Conditions and Sample Preparation
3.2. Metabolite Profiling and Quantification
3.3. Multivariate Data Analysis
3.4. Yeast Chronological Lifespan Measurement
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jia, Y.; Rothermel, B.; Thornton, J.; Butow, R.A. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol. Cell. Biol. 1997, 17, 1110–1117. [Google Scholar]
- Liao, X.; Butow, R.A. RTG1 and RTG2: Two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 1993, 72, 61–71. [Google Scholar]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: the retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Liu, Z.; Butow, R.A. Mitochondrial retrograde signaling. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef]
- Sekito, T.; Thornton, J.; Butow, R.A. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol. Biol. Cell 2000, 11, 2103–2115. [Google Scholar] [CrossRef]
- Rothermel, B.A.; Thornton, J.L.; Butow, R.A. Rtg3p, a basic helix-loop-helix/leucine zipper protein that functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J. Biol. Chem. 1997, 272, 19801–19807. [Google Scholar] [CrossRef]
- Chelstowska, A.; Butow, R.A. RTG genes in yeast that function in communication between mitochondria and the nucleus are also required for expression of genes encoding peroxisomal proteins. J. Biol. Chem. 1995, 270, 18141–18146. [Google Scholar] [CrossRef]
- Rothermel, B.A.; Shyjan, A.W.; Etheredge, J.L.; Butow, R.A. Transactivation by Rtg1p, a basic helix-loop-helix protein that functions in communication between mitochondria and the nucleus in yeast. J. Biol. Chem. 1995, 270, 29476–29482. [Google Scholar] [CrossRef]
- Traven, A.; Wong, J.M.; Xu, D.; Sopta, M.; Ingles, C.J. Interorganellar communication. Altered nuclear gene expression profiles in a yeast mitochondrial DNA mutant. J. Biol. Chem. 2001, 276, 4020–4027. [Google Scholar]
- Komeili, A.; Wedaman, K.P.; O’Shea, E.K.; Powers, T. Mechanism of metabolic control: Target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J. Cell Biol. 2000, 151, 863–878. [Google Scholar] [CrossRef]
- Tate, J.J.; Cox, K.H.; Rai, R.; Cooper, T.G. Mks1p is required for negative regulation of retrograde gene expression in Saccharomyces cerevisiae but does not affect nitrogen catabolite repression-sensitive gene expression. J. Biol. Chem. 2002, 277, 20477–20482. [Google Scholar] [CrossRef]
- Crespo, J.L.; Powers, T.; Fowler, B.; Hall, M.N. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc. Natl. Acad. Sci. USA 2002, 99, 6784–6789. [Google Scholar] [CrossRef]
- Dilova, I.; Chen, C.-Y.; Powers, T. Mks1 in concert with TOR signaling negatively regulates RTG target gene expression in S. cerevisiae. Curr. Biol. 2002, 12, 389–395. [Google Scholar]
- Dilova, I.; Aronova, S.; Chen, J.C.-Y.; Powers, T. Tor signaling and nutrient-based signals converge on Mks1p phosphorylation to regulate expression of Rtg1.Rtg3p-dependent target genes. J. Biol. Chem. 2004, 279, 46527–46535. [Google Scholar] [CrossRef]
- Kirchman, P.A.; Kim, S.; Lai, C.-Y.; Jazwinski, S.M. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 1999, 152, 179–190. [Google Scholar]
- Liu, Z.; Butow, R.A. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol. Cell. Biol. 1999, 19, 6720–6728. [Google Scholar]
- Fiehn, O. Metabolomics–the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Fukusaki, E.; Kobayashi, A. Plant metabolomics: potential for practical operation. J. Biosci. Bioeng. 2005, 100, 347–354. [Google Scholar] [CrossRef]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 2001, 19, 45–50. [Google Scholar] [CrossRef]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003, 21, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, R.; Tamura, T.; Takaoka, C.; Harada, K.; Kobayashi, A.; Mukai, Y.; Fukusaki, E. Metabolomics-based systematic prediction of yeast lifespan and its application for semi-rational screening of ageing-related mutants. Aging Cell 2010, 9, 616–625. [Google Scholar] [CrossRef]
- Shirai, T.; Matsuda, F.; Okamoto, M.; Kondo, A. Evaluation of control mechanisms for Saccharomyces cerevisiae central metabolic reactions using metabolome data of eight single-gene deletion mutants. Appl. Microbiol. Biotechnol. 2013, 97, 3569–3577. [Google Scholar] [CrossRef]
- Hashim, Z.; Teoh, S.T.; Bamba, T.; Fukusaki, E. Construction of a metabolome library for transcription factor-related single gene mutants of Saccharomyces cerevisiae. J. Chromatogr. B. 2014. Available online: http://dx.doi.org/10.1016/j.jchromb.2014.05.041.
- Gika, H.G.; Theodoridis, G.A.; Wingate, J.E.; Wilson, I.D. Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: application to human urine. J. Proteome Res. 2007, 6, 3291–3303. [Google Scholar] [CrossRef]
- Zelena, E.; Dunn, W.B.; Broadhurst, D.; Francis-McIntyre, S.; Carroll, K.M.; Begley, P.; O’Hagan, S.; Knowles, J.D.; Halsall, A.; Wilson, I.D.; et al. Development of a robust and repeatable UPLC-MS method for the long-term metabolomic study of human serum. Anal. Chem. 2009, 81, 1357–1364. [Google Scholar] [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Earll, M.; Wilson, I.D. A QC approach to the determination of day-to-day reproducibility and robustness of LC-MS methods for global metabolite profiling in metabonomics/metabolomics. Bioanalysis 2012, 4, 2239–2247. [Google Scholar] [CrossRef]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. MetaboAnalyst 2.0–A comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar] [CrossRef]
- Poyton, R.O.; McEwen, J.E. Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 1996, 65, 563–607. [Google Scholar] [CrossRef]
- Zampar, G.G.; Kümmel, A.; Ewald, J.; Jol, S.; Niebel, B.; Picotti, P.; Aebersold, R.; Sauer, U.; Zamboni, N.; Heinemann, M. Temporal system-level organization of the switch from glycolytic to gluconeogenic operation in yeast. Mol. Syst. Biol. 2013, 9, 651. [Google Scholar]
- DeRisi, J.L.; Iyer, V.R.; Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997, 278, 680–686. [Google Scholar] [CrossRef]
- Eisenberg, T.; Carmona-Gutierrez, D.; Büttner, S.; Tavernarakis, N.; Madeo, F. Necrosis in yeast. Apoptosis 2010, 15, 257–268. [Google Scholar] [CrossRef]
- Farrugia, G.; Balzan, R. Oxidative stress and programmed cell death in yeast. Front. Oncol. 2012, 2, 2–21. [Google Scholar]
- Small, W.C.; Brodeur, R.D.; Sandor, A.; Fedorova, N.; Li, G.; Butow, R.A.; Srere, P.A. Enzymic and metabolic studies on retrograde regulation mutants of yeast. Biochemistry 1995, 34, 5569–5576. [Google Scholar] [CrossRef]
- Kemmeren, P.; Sameith, K.; van de Pasch, L.A.L.; Benschop, J.J.; Lenstra, T.L.; Margaritis, T.; O’Duibhir, E.; Apweiler, E.; van Wageningen, S.; Ko, C.W.; et al. Large-scale genetic perturbationsreveal regulatory networks and an abundance of gene-specific repressors. Cell 2014, 157, 740–752. [Google Scholar]
- Grenson, M.; Hou, C.; Crabeel, M. Multiplicity of the amino acid permeases in Saccharomyces cerevisiae IV. Evidence for a general amino acid permease. J. Bacteriol. 1970, 103, 770–777. [Google Scholar]
- Jauniaux, J.-C.; Grenson, M. GAP1, the general amino acid permease gene of Saccharomyces cerevisiae. Nucleotide sequence, protein similarity with the other bakers yeast amino acid permeases, and nitrogen catabolite repression. Eur. J. Biochem. 1990, 190, 39–44. [Google Scholar] [CrossRef]
- Chen, E.J.; Kaiser, C.A. Amino acids regulate the intracellular trafficking of the general amino acid permease of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 14837–14842. [Google Scholar] [CrossRef]
- Schreve, J.L.; Sin, J.K.; Garrett, J.M. The Saccharomyces cerevisiae YCC5 (YCL025c) gene encodes an amino acid permease, Agp1, which transports asparagine and glutamine. J. Bacteriol. 1998, 180, 2556–2559. [Google Scholar]
- Borghouts, C.; Benguria, A.; Wawryn, J.; Jazwinski, S.M. Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics 2004, 166, 765–777. [Google Scholar] [CrossRef]
- Barros, M.H.; Bandy, B.; Tahara, E.B.; Kowaltowski, A.J. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 49883–49888. [Google Scholar] [CrossRef]
- Powers, R.W.; Kaeberlein, M.; Caldwell, S.D.; Kennedy, B.K.; Fields, S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006, 20, 174–184. [Google Scholar] [CrossRef]
- Fabrizio, P.; Longo, V.D. The chronological life span of Saccharomyces cerevisiae. Aging Cell 2003, 2, 73–81. [Google Scholar] [CrossRef]
- Alvers, A.L.; Fishwick, L.K.; Wood, M.S.; Hu, D.; Chung, H.S.; Dunn, W.A.; Aris, J.P. Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 2009, 8, 353–369. [Google Scholar] [CrossRef]
- Kaiser, C.; Michaelis, S.; Mitchell, A. Methods in yeast genetics: A Cold Spring Harbor Laboratory Course Manual; Cold Spring Harbor: New York, NY, USA, 1994; p. 234. [Google Scholar]
- De Hoon, M.J.L.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef]
- Saldanha, A.J. Java Treeview–extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef]
- Klukas, C.; Schreiber, F. Integration of -omics data and networks for biomedical research with VANTED. J. Integr. Bioinform. 2010, 7, 112–118. [Google Scholar]
- Parrella, E.; Longo, V.D. The chronological life span of Saccharomyces cerevisiae to study mitochondrial dysfunction and disease. Methods 2008, 46, 256–262. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hashim, Z.; Mukai, Y.; Bamba, T.; Fukusaki, E. Metabolic Profiling of Retrograde Pathway Transcription Factors Rtg1 and Rtg3 Knockout Yeast. Metabolites 2014, 4, 580-598. https://doi.org/10.3390/metabo4030580
Hashim Z, Mukai Y, Bamba T, Fukusaki E. Metabolic Profiling of Retrograde Pathway Transcription Factors Rtg1 and Rtg3 Knockout Yeast. Metabolites. 2014; 4(3):580-598. https://doi.org/10.3390/metabo4030580
Chicago/Turabian StyleHashim, Zanariah, Yukio Mukai, Takeshi Bamba, and Eiichiro Fukusaki. 2014. "Metabolic Profiling of Retrograde Pathway Transcription Factors Rtg1 and Rtg3 Knockout Yeast" Metabolites 4, no. 3: 580-598. https://doi.org/10.3390/metabo4030580
APA StyleHashim, Z., Mukai, Y., Bamba, T., & Fukusaki, E. (2014). Metabolic Profiling of Retrograde Pathway Transcription Factors Rtg1 and Rtg3 Knockout Yeast. Metabolites, 4(3), 580-598. https://doi.org/10.3390/metabo4030580