Limitations of Deuterium-Labelled Substrates for Quantifying NADPH Metabolism in Heterotrophic Arabidopsis Cell Cultures

 ,
, 
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Deuterium Labelling in Vitro
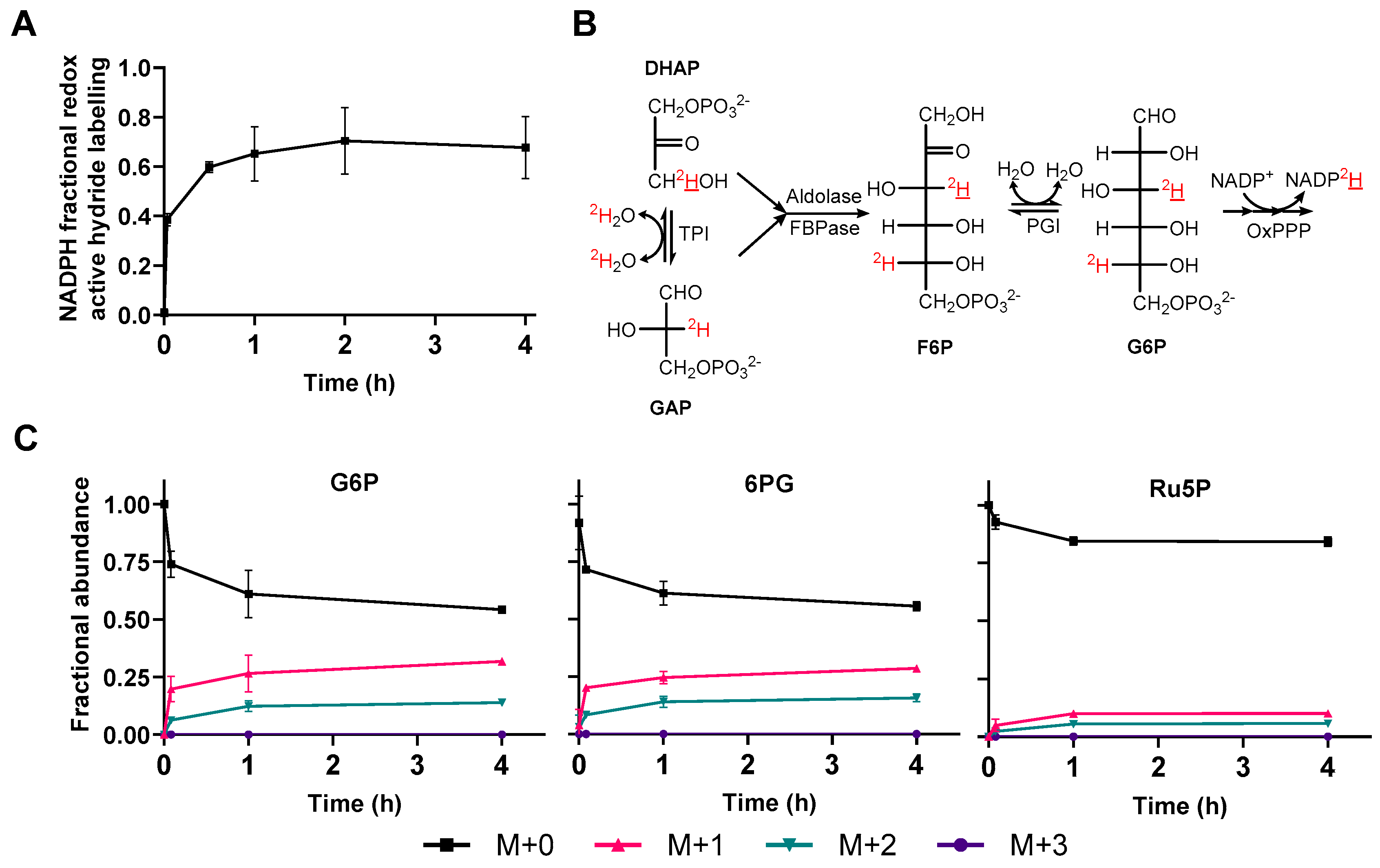
2.2. Water Exchange in Vivo
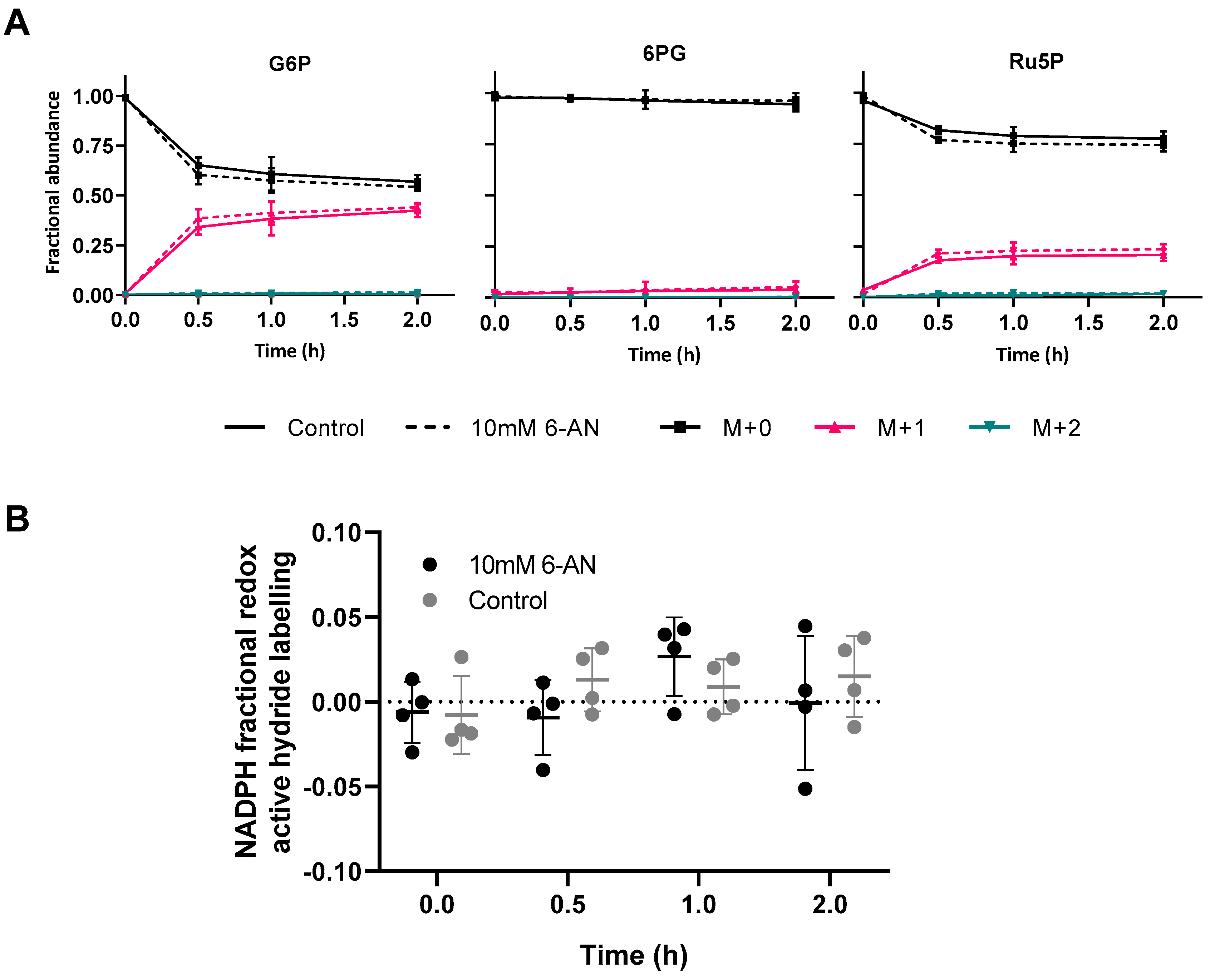
2.3. [1-2H]Glucose Labelling in Vivo
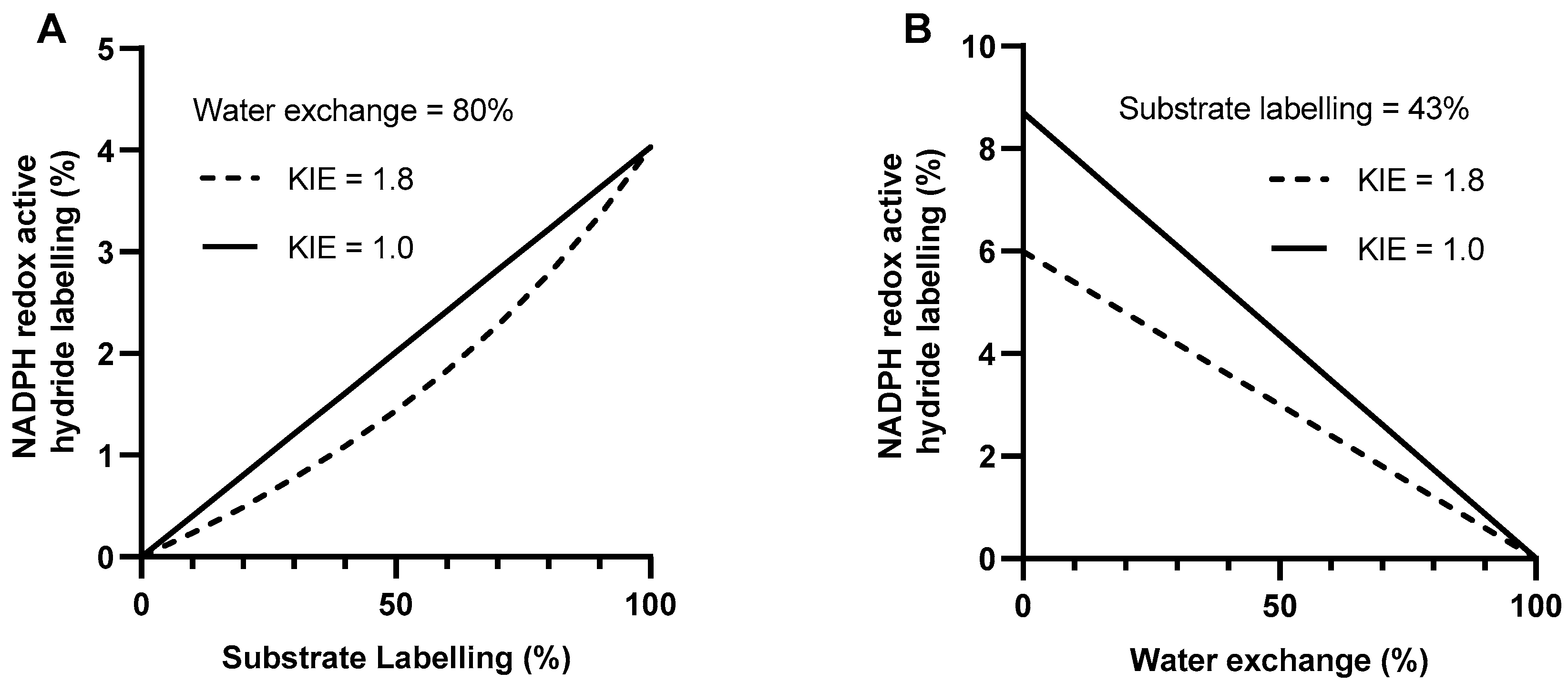
2.4. Simulating NADPH Redox Active Hydride Labelling
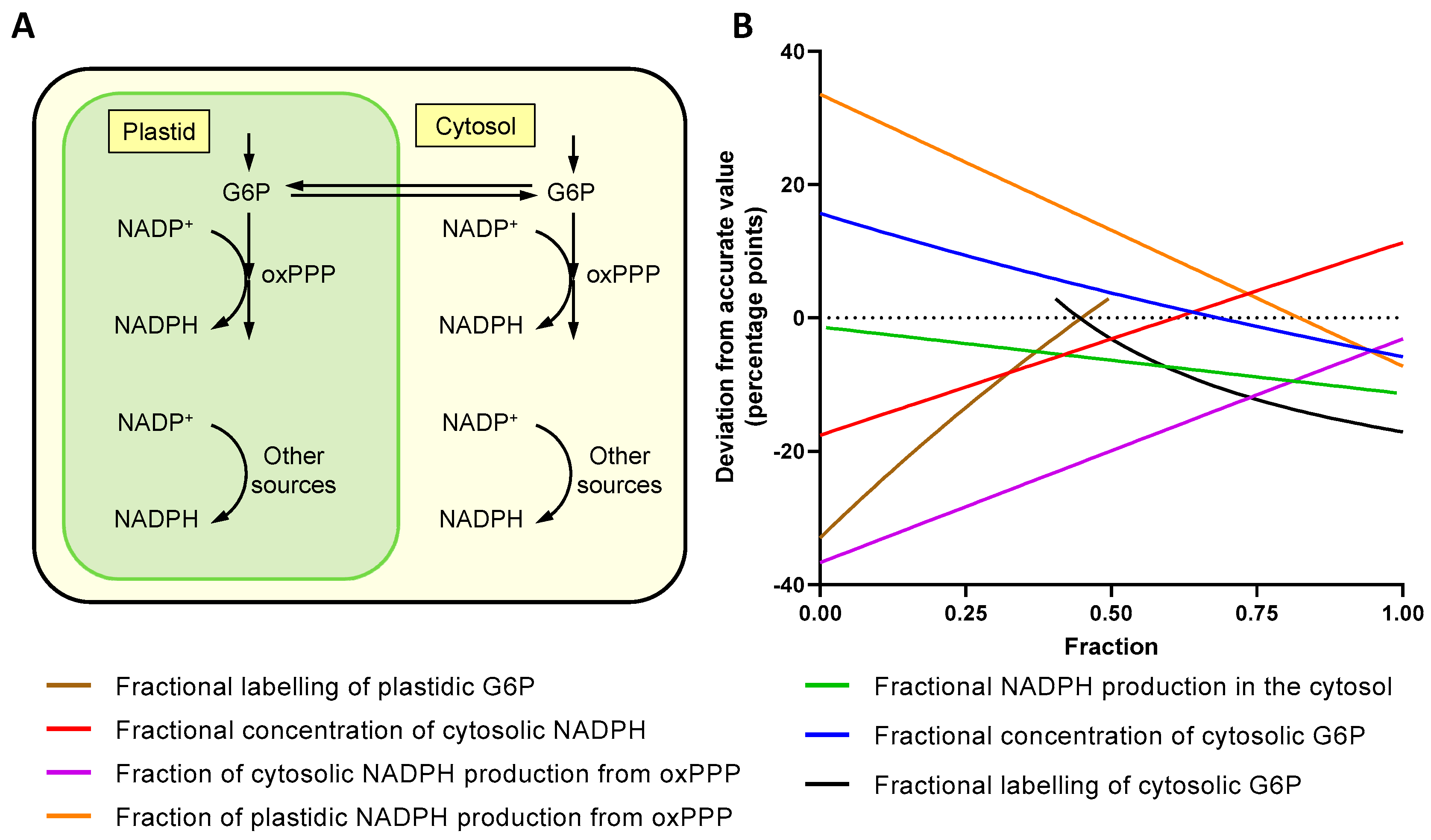
2.5. The Effect of Subcellular Compartmentation
3. Discussion
3.1. Flavin-Enzyme-Catalysed Water Exchange Abolishes Detectable Labelling from [1-2H]glucose
3.2. Low Substrate Labelling Decreases Maximum Possible Labelling Signal
3.3. Metabolic Network Structure Prevents Accurate Measurement
4. Conclusions
5. Materials and Methods
5.1. Arabidopsis Cell Culture
5.2. Metabolite Extraction
5.3. Liquid Chromatography Mass Spectrometry
5.4. Simulating NADPH Redox Active Hydride Labelling
5.5. Redox Active Hydride Labelling
5.6. In Vitro oxPPP Labelling
5.7. In Vitro NADPH–Water Exchange by Glutathione Reductase
5.8. In Vivo D2O Labelling
5.9. In Vivo [1-2H]glucose Labelling and 6-AN Treatment
5.10. Proteomic Analysis of Cell Extract
5.11. PaxDB Quantitative Proteome Analysis
5.12. Compartmentation Simulation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hashida, S.N.; Takahashi, H.; Uchimiya, H. The role of NAD biosynthesis in plant development and stress responses. Ann. Bot. 2009, 103, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noctor, G.; Queval, G.; Gakière, B. NAD(P) synthesis and pyridine nucleotide cycling in plants and their potential importance in stress conditions. J. Exp. Bot. 2006, 57, 1603–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gakière, B.; Fernie, A.R.; Pétriacq, P. More to NAD+ than meets the eye: A regulator of metabolic pools and gene expression in Arabidopsis. Free Radic. Biol. Med. 2018, 122, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Arrivault, S.; Guenther, M.; Ivakov, A.; Feil, R.; Vosloh, D.; Van Dongen, J.T.; Sulpice, R.; Stitt, M. Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 2009, 59, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Gorelova, V.; De Lepeleire, J.; Van Daele, J.; Pluim, D.; Meï, C.; Cuypers, A.; Leroux, O.; Rébeillé, F.; Schellens, J.; Blancquaert, D.; et al. Dihydrofolate reductase/thymidylate synthase fine-tunes the folate status and controls redox homeostasis in plants. Plant Cell 2017, 29, 2831–2853. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.F.; Chen, Q.J.; An, R.; Chen, Y.M.; Chen, J.; Wang, X.C. NADK2, an Arabidopsis chloroplastic NAD kinase, plays a vital role in both chlorophyll synthesis and chloroplast protection. Plant Mol. Biol. 2005, 59, 553–564. [Google Scholar] [CrossRef]
- Chai, M.F.; Wei, P.C.; Chen, Q.J.; An, R.; Chen, J.; Yang, S.; Wang, X.C. NADK3, a novel cytoplasmic source of NADPH, is required under conditions of oxidative stress and modulates abscisic acid responses in Arabidopsis. Plant J. 2006, 47, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Turner, W.L.; Waller, J.C.; Vanderbeld, B.; Snedden, W.A. Cloning and characterization of two NAD kinases from Arabidopsis. Identification of a calmodulin binding isoform. Plant Physiol. 2004, 135, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Voll, L.M.; Zell, M.B.; Engelsdorf, T.; Saur, A.; Wheeler, M.G.; Drincovich, M.F.; Weber, A.P.M.; Maurino, V.G. Loss of cytosolic NADP-malic enzyme 2 in Arabidopsis thaliana is associated with enhanced susceptibility to Colletotrichum higginsianum. New Phytol. 2012, 195, 189–202. [Google Scholar] [CrossRef]
- Mhamdi, A.; Mauve, C.; Gouia, H.; Saindrenan, P.; Hodges, M.; Noctor, G. Cytosolic NADP-dependent isocitrate dehydrogenase contributes to redox homeostasis and the regulation of pathogen responses in Arabidopsis leaves. Plant Cell Environ. 2010, 33, 1112–1123. [Google Scholar]
- Mhamdi, A.; Noctor, G. Analysis of the roles of the Arabidopsis peroxisomal isocitrate dehydrogenase in leaf metabolism and oxidative stress. Environ. Exp. Bot. 2015, 114, 22–29. [Google Scholar] [CrossRef]
- Wakao, S.; Andre, C.; Benning, C. Functional analyses of cytosolic glucose-6-phosphate dehydrogenases and their contribution to seed oil accumulation in Arabidopsis. Plant Physiol. 2008, 146, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Corpas, F.J.; Barroso, J.B. NADPH-generating dehydrogenases: Their role in the mechanism of protection against nitro-oxidative stress induced by adverse environmental conditions. Front. Environ. Sci. 2014, 2, 55. [Google Scholar] [CrossRef]
- Chen, X.; Li, S.; Liu, L. Engineering redox balance through cofactor systems. Trends Biotechnol. 2014, 32, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; San, K.Y.; Bennett, G.N. Cofactor engineering for advancing chemical biotechnology. Curr. Opin. Biotechnol. 2013, 24, 994–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, K.; Wasylenko, T.M.; Zhou, K.; Xu, P.; Stephanopoulos, G. lipid production inYarrowia lipolytica maximized by engineering cytosolic redox metabolism. Nat. Biotechnol. 2017, 35, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.J.; Xia, Z.F.; Fu, N.H.; Nie, Z.K.; Shen, M.Q.; Tian, Q.Q.; Huang, H. Cofactor engineering through heterologous expression of an NADH oxidase and its impact on metabolic flux redistribution in Klebsiella pneumoniae. Biotechnol. Biofuels 2013, 6, 7. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.; Zhou, P.; Huang, J.; Xia, X.; Li, W.; Zhou, Z.; Chen, Y.; Liu, Y.; Dong, M. Improving metabolic efficiency of the reverse beta-oxidation cycle by balancing redox cofactor requirement. Metab. Eng. 2017, 44, 313–324. [Google Scholar] [CrossRef]
- Sánchez, A.M.; Andrews, J.; Hussein, I.; Bennett, G.N.; San, K.Y. Effect of overexpression of a soluble pyridine nucleotide transhydrogenase (UdhA) on the production of poly(3-hydroxybutyrate) in Escherichia coli. Biotechnol. Prog. 2006, 22, 420–425. [Google Scholar] [CrossRef]
- Martínez, I.; Zhu, J.; Lin, H.; Bennett, G.N.; San, K.Y. Replacing Escherichia coli NAD-dependent glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with a NADP-dependent enzyme from Clostridium acetobutylicum facilitates NADPH dependent pathways. Metab. Eng. 2008, 10, 352–359. [Google Scholar] [CrossRef]
- Chemler, J.A.; Fowler, Z.L.; McHugh, K.P.; Koffas, M.A.G. Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab. Eng. 2010, 12, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, Q.; Li, Y.; Zhang, Y. Engineering redox homeostasis to develop efficient alcohol-producing microbial cell factories. Microb. Cell Fact. 2017, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.M.; Evans, J.R. The importance of energy balance in improving photosynthetic productivity. Plant Physiol. 2011, 155, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G.; Hodges, M. Respiration and nitrogen assimilation: Targeting mitochondria-associated metabolism as a means to enhance nitrogen use efficiency. J. Exp. Bot. 2011, 62, 1467–1482. [Google Scholar] [CrossRef] [PubMed]
- Zinta, G.; Khan, A.; AbdElgawad, H.; Verma, V.; Srivastava, A.K. Unveiling the redox control of plant reproductive development during abiotic stress. Front. Plant Sci. 2016, 7, 700. [Google Scholar] [CrossRef] [PubMed]
- Scharte, J.; Schön, H.; Tjaden, Z.; Weis, E.; von Schaewen, A. Isoenzyme replacement of glucose-6-phosphate dehydrogenase in the cytosol improves stress tolerance in plants. Proc. Natl. Acad. Sci. USA 2009, 106, 8061–8066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masakapalli, S.K.; Le Lay, P.; Huddleston, J.E.; Pollock, N.L.; Kruger, N.J.; Ratcliffe, R.G. Subcellular flux analysis of central metabolism in a heterotrophic Arabidopsis cell suspension using steady-state stable isotope labeling. Plant Physiol. 2010, 152, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Masakapalli, S.K.; Kruger, N.J.; Ratcliffe, R.G. The metabolic flux phenotype of heterotrophic Arabidopsis cells reveals a complex response to changes in nitrogen supply. Plant J. 2013, 74, 569–582. [Google Scholar] [CrossRef]
- Masakapalli, S.K.; Bryant, F.M.; Kruger, N.J.; Ratcliffe, R.G. The metabolic flux phenotype of heterotrophic Arabidopsis cells reveals a flexible balance between the cytosolic and plastidic contributions to carbohydrate oxidation in response to phosphate limitation. Plant J. 2014, 78, 964–977. [Google Scholar] [CrossRef]
- Suarez-Mendez, C.A.; Hanemaaijer, M.; ten Pierick, A.; Wolters, J.C.; Heijnen, J.J.; Wahl, S.A. Interaction of storage carbohydrates and other cyclic fluxes with central metabolism: A quantitative approach by non-stationary 13C metabolic flux analysis. Metab. Eng. Commun. 2016, 3, 52–63. [Google Scholar] [CrossRef]
- Cheung, C.Y.M.; Williams, T.C.R.; Poolman, M.G.; Fell, D.A.; Ratcliffe, R.G.; Sweetlove, L.J. A method for accounting for maintenance costs in flux balance analysis improves the prediction of plant cell metabolic phenotypes under stress conditions. Plant J. 2013, 75, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shah, S.; Fan, J.; Park, J.O.; Wellen, K.E.; Rabinowitz, J.D. Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat. Chem. Biol. 2016, 12, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, L.; Liu, L.; Su, X.; Rabinowitz, J.D. Chemical basis for deuterium labeling of fat and NADPH. J. Am. Chem. Soc. 2017, 139, 14368–14371. [Google Scholar] [CrossRef]
- Kazuki, S.; Akihiko, K.; Shigenobu, O.; Yousuke, S.; Tamio, Y. Incorporation of hydrogen atoms from deuterated water and stereospecifically deuterium-labeled nicotinamide nucleotides into fatty acids with the Escherichia coli fatty acid synthetase system. Biochim. Biophys. Acta 1980, 618, 202–213. [Google Scholar] [CrossRef]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef]
- Fletcher, S.J.; Herlihy, J.M.; Knowles, J.R.; Albery, W.J. Energetics of triosephosphate isomerase: The appearance of solvent tritium in substrate glyceraldehyde 3-phosphate and in product. Biochemistry 1976, 15, 5612–5617. [Google Scholar] [CrossRef]
- Maister, S.G.; Pett, C.P.; Knowles, J.R.; Albery, W.J. Energetics of triosephosphate isomerase: The appearance of solvent tritium in substrate dihydroxyacetone phosphate and in product. Biochemistry 1976, 15, 5607–5612. [Google Scholar] [CrossRef]
- Schleucher, J.; Vanderveer, P.; Markley, J.L.; Sharkey, T.D. Intramolecular deuterium distributions reveal disequilibrium of chloroplast phosphoglucose isomerase. Plant Cell Environ. 1999, 22, 525–533. [Google Scholar] [CrossRef]
- Fernie, A.R.; Roscher, A.; Ratcliffe, R.G.; Kruger, N.J. Fructose 2,6-bisphosphate activates pyrophosphate: Fructose-6-phosphate 1-phosphotransferase and increases triose phosphate to hexose phosphate cycling in heterotrophic cells. Planta 2001, 212, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Letisse, F.; Sokol, S.; Portais, J.C. IsoCor: Correcting MS data in isotope labeling experiments. Bioinformatics 2012, 28, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Garlick, A.P.; Moore, C.; Kruger, N.J. Monitoring flux through the oxidative pentose phosphate pathway using [1-14C]gluconate. Planta 2002, 216, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Köhler, E.; Barrach, H.J.; Neubert, D. Inhibition of NADP dependent oxidoreductases by the 6-aminonicotinamide analogue of NADP. FEBS Lett. 1970, 6, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Gakière, B.; Hao, J.; de Bont, L.; Pétriacq, P.; Nunes-Nesi, A.; Fernie, A.R. NAD+ biosynthesis and signaling in plants. CRC. Crit. Rev. Plant Sci. 2018, 37, 259–307. [Google Scholar] [CrossRef]
- Wakao, S.; Benning, C. Genome-wide analysis of glucose-6-phosphate dehydrogenases in Arabidopsis. Plant J. 2005, 41, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Heise, R.; Fernie, A.R.; Stitt, M.; Nikoloski, Z. Pool size measurements facilitate the determination of fluxes at branching points in non-stationary metabolic flux analysis: The case of Arabidopsis thaliana. Front. Plant Sci. 2015, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Igamberdiev, A.U.; Gardeström, P. Regulation of NAD- and NADP-dependent isocitrate dehydrogenases by reduction levels of pyridine nucleotides in mitochondria and cytosol of pea leaves. Biochim. Biophys. Acta 2003, 1606, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Heineke, D.; Riens, B.; Grosse, H.; Hoferichter, P.; Peter, U.; Flugge, U.; Heldt, H.W. Redox transfer across the inner chloroplast envelope membrane. Plant Physiol. 1991, 95, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yoseph, O.; Kingsley, P.B.; Ross, B.D. Metabolic loss of deuterium from isotopically labeled glucose. Magn. Reson. Med. 1994, 32, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shreve, D.S.; Levy, H.R. Kinetic mechanism of glucose-6-phosphate dehydrogenase from the lactating rat mammary gland. Implications for regulation. J. Biol. Chem. 1980, 255, 2670–2677. [Google Scholar] [PubMed]
- Ma, F.; Jazmin, L.J.; Young, J.D.; Allen, D.K. Isotopically nonstationary 13C flux analysis of changes in Arabidopsis thaliana leaf metabolism due to high light acclimation. Proc. Natl. Acad. Sci. USA 2014, 111, 16967–16972. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, R.; Stitt, M.; Heldt, H.W. Subcellular metabolite levels in spinach leaves: Regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiol. 1987, 83, 399–407. [Google Scholar] [CrossRef]
- Szecowka, M.; Heise, R.; Tohge, T.; Nunes-Nesi, A.; Vosloh, D.; Huege, J.; Feil, R.; Lunn, J.; Nikoloski, Z.; Stitt, M.; et al. Metabolic fluxes in an illuminated Arabidopsis rosette. Plant Cell 2013, 25, 694–714. [Google Scholar] [CrossRef] [PubMed]
- Fuhrer, T.; Sauer, U. Different biochemical mechanisms ensure network-wide balancing of reducing equivalents in microbial metabolism. J. Bacteriol. 2009, 191, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.R.; Miguet, L.; Masakapalli, S.K.; Kruger, N.J.; Sweetlove, L.J.; Ratcliffe, R.G. Metabolic network fluxes in heterotrophic Arabidopsis cells: Stability of the flux distribution under different oxygenation conditions. Plant Physiol. 2008, 148, 704–18. [Google Scholar] [CrossRef]
- Horsch, R.B.; King, J.; Jones, G.E. Measurement of cultured plant cell growth on filter paper discs. Can. J. Bot. 1980, 58, 2402–2406. [Google Scholar] [CrossRef]
- Scholten, H.J. A plating technique for poorly growing cell cultures of Arabidopsis thaliana. Arab. Inf. Serv. 1980, 17, 27. [Google Scholar]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh performance liquid chromatography-tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, E.N.; McCullagh, J.S.O.; Ratcliffe, R.G.; Kruger, N.J. Limitations of Deuterium-Labelled Substrates for Quantifying NADPH Metabolism in Heterotrophic Arabidopsis Cell Cultures. Metabolites 2019, 9, 205. https://doi.org/10.3390/metabo9100205
Smith EN, McCullagh JSO, Ratcliffe RG, Kruger NJ. Limitations of Deuterium-Labelled Substrates for Quantifying NADPH Metabolism in Heterotrophic Arabidopsis Cell Cultures. Metabolites. 2019; 9(10):205. https://doi.org/10.3390/metabo9100205
Chicago/Turabian StyleSmith, Edward N., James S. O. McCullagh, R. George Ratcliffe, and Nicholas J. Kruger. 2019. "Limitations of Deuterium-Labelled Substrates for Quantifying NADPH Metabolism in Heterotrophic Arabidopsis Cell Cultures" Metabolites 9, no. 10: 205. https://doi.org/10.3390/metabo9100205
APA StyleSmith, E. N., McCullagh, J. S. O., Ratcliffe, R. G., & Kruger, N. J. (2019). Limitations of Deuterium-Labelled Substrates for Quantifying NADPH Metabolism in Heterotrophic Arabidopsis Cell Cultures. Metabolites, 9(10), 205. https://doi.org/10.3390/metabo9100205