Glucose-6 Phosphate, a Central Hub for Liver Carbohydrate Metabolism

Abstract
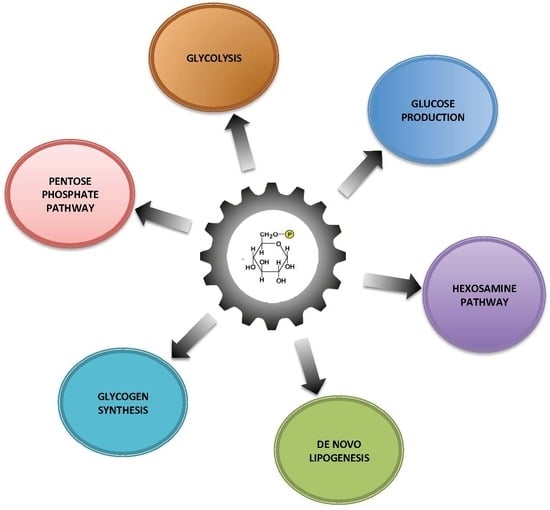
:
{kind=link}
{kind=link}
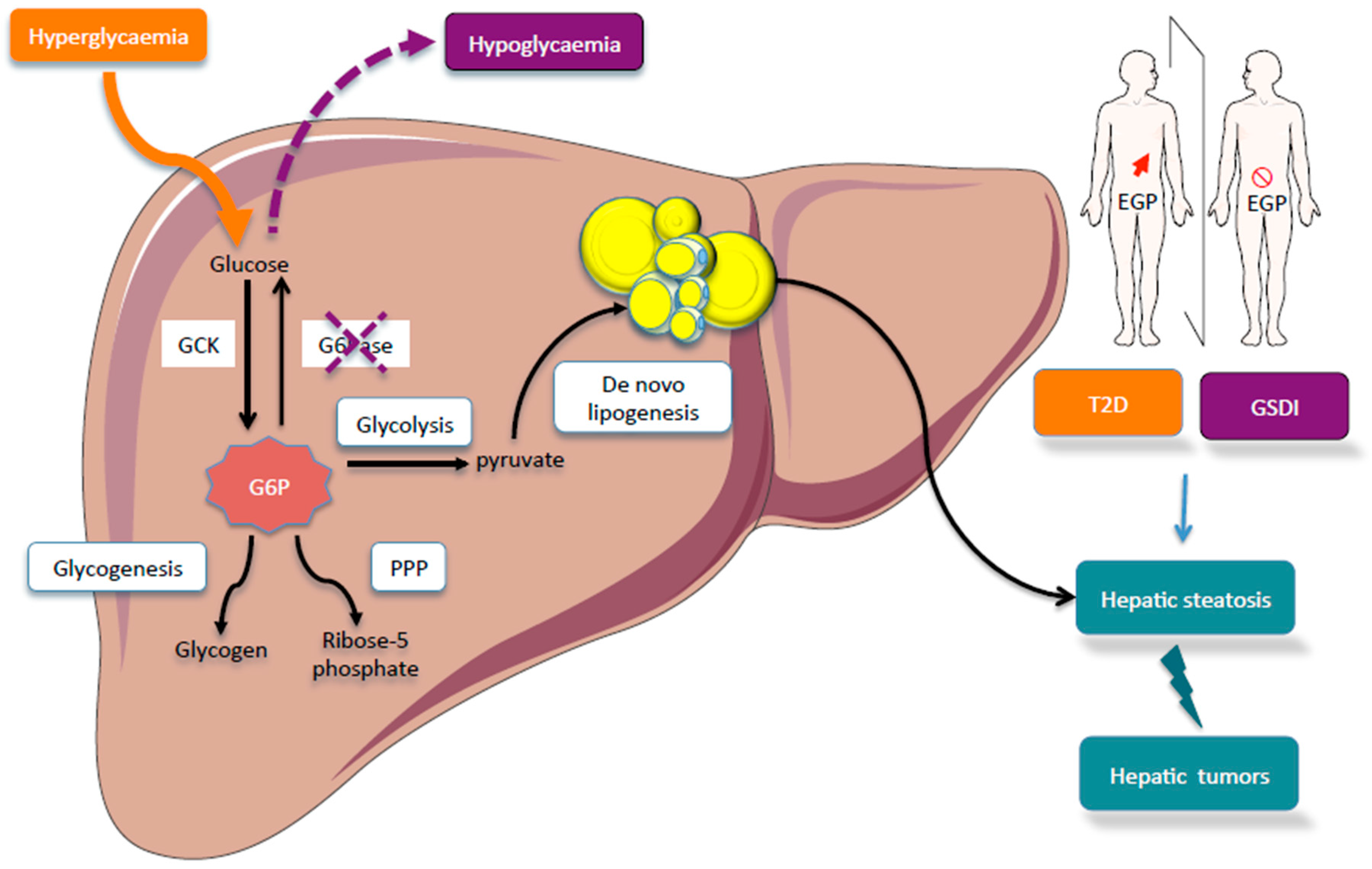{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Metabolic Fate of Glucose-6 Phosphate in the Healthy Liver
2.1. Glucose and Lipid Storage
2.2. Maintenance of Glycaemia and Endogenous Glucose Production
2.3. Glucose-6 Phosphate: A Source of Energy and Carbon Skeletons
2.4. Hexosamine Pathway
3. ChREBP: A Glucose Sensor
4. Imbalance of Glucose-6 Phosphate Metabolism Leads to Metabolic Diseases and Promotes Hepatocarcinogenesis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Schaftingen, E.; Detheux, M.; Veiga da Cunha, M. Short-term control of glucokinase activity: Role of a regulatory protein. FASEB J. 1994, 8, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Agius, L. Hormonal and Metabolite Regulation of Hepatic Glucokinase. Annu. Rev. Nutr. 2016, 36, 389–415. [Google Scholar] [CrossRef] [PubMed]
- Baltrusch, S.; Lenzen, S.; Okar, D.A.; Lange, A.J.; Tiedge, M. Characterization of glucokinase-binding protein epitopes by a phage-displayed peptide library identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel interaction partner. J. Biol. Chem. 2001, 276, 43915–43923. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, B.S.; Boss, A.; van Dijk, T.H.; Soty, M.; Wolters, H.; Mutel, E.; Groen, A.K.; Derks, T.G.J.; Mithieux, G.; Heerschap, A.; et al. Hepatocytes contribute to residual glucose production in a mouse model for glycogen storage disease type Ia. Hepatology 2017, 66, 2042–2054. [Google Scholar] [CrossRef]
- Rajas, F.; Labrune, P.; Mithieux, G. Glycogen storage disease type 1 and diabetes: Learning by comparing and contrasting the two disorders. Diabetes Metab. 2013, 39, 377–387. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen metabolism in humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Magnusson, I.; Rothman, D.L.; Jucker, B.; Cline, G.W.; Shulman, R.G.; Shulman, G.I. Liver glycogen turnover in fed and fasted humans. Am. J. Physiol. 1994, 266, E796–E803. [Google Scholar] [CrossRef]
- McDevitt, R.M.; Bott, S.J.; Harding, M.; Coward, W.A.; Bluck, L.J.; Prentice, A.M. De novo lipogenesis during controlled overfeeding with sucrose or glucose in lean and obese women. Am. J. Clin. Nutr. 2001, 74, 737–746. [Google Scholar] [CrossRef]
- Ozen, H. Glycogen storage diseases: New perspectives. World J. Gastroenterol. 2007, 13, 2541–2553. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef]
- Xu, K.; Morgan, K.T.; Todd Gehris, A.; Elston, T.C.; Gomez, S.M. A Whole-Body Model for Glycogen Regulation Reveals a Critical Role for Substrate Cycling in Maintaining Blood Glucose Homeostasis. PLoS Comput. Biol. 2011, 7, e1002272. [Google Scholar] [CrossRef] [PubMed]
- Mutel, E.; Gautier-Stein, A.; Abdul-Wahed, A.; Amigó-Correig, M.; Zitoun, C.; Stefanutti, A.; Houberdon, I.; Tourette, J.A.; Mithieux, G.; Rajas, F. Control of Blood Glucose in the Absence of Hepatic Glucose Production During Prolonged Fasting in Mice. Diabetes 2011, 60, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Mithieux, G. New data and concepts on glutamine and glucose metabolism in the gut. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Mithieux, G.; Rajas, F.; Gautier-Stein, A. A novel role for glucose 6-phosphatase in the small intestine in the control of glucose homeostasis. J. Biol. Chem. 2004, 279, 44231–44234. [Google Scholar] [CrossRef] [PubMed]
- Soty, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Gut-Brain Glucose Signaling in Energy Homeostasis. Cell Metab. 2017, 25, 1231–1242. [Google Scholar] [CrossRef]
- Pillot, B.; Soty, M.; Gautier-Stein, A.; Zitoun, C.; Mithieux, G. Protein feeding promotes redistribution of endogenous glucose production to the kidney and potentiates its suppression by insulin. Endocrinology 2009, 150, 616–624. [Google Scholar] [CrossRef]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef]
- Croset, M.; Rajas, F.; Zitoun, C.; Hurot, J.M.; Montano, S.; Mithieux, G. Rat small intestine is an insulin-sensitive gluconeogenic organ. Diabetes 2001, 50, 740–746. [Google Scholar] [CrossRef]
- Mithieux, G.; Gautier-Stein, A.; Rajas, F.; Zitoun, C. Contribution of intestine and kidney to glucose fluxes in different nutritional states in rat. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2006, 143, 195–200. [Google Scholar] [CrossRef]
- Mutel, E.; Abdul-Wahed, A.; Ramamonjisoa, N.; Stefanutti, A.; Houberdon, I.; Cavassila, S.; Pilleul, F.; Beuf, O.; Gautier-Stein, A.; Penhoat, A.; et al. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J. Hepatol. 2011, 54, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Penhoat, A.; Fayard, L.; Stefanutti, A.; Mithieux, G.; Rajas, F. Intestinal gluconeogenesis is crucial to maintain a physiological fasting glycemia in the absence of hepatic glucose production in mice. Metab. Clin. Exp. 2014, 63, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Morris, H.P.; Weber, G. Behavior of transaldolase (EC 2.2.1.2) and transketolase (EC 2.2.1.1) Activities in normal, neoplastic, differentiating, and regenerating liver. Cancer Res. 1976, 36, 3189–3197. [Google Scholar] [PubMed]
- Hanczko, R.; Fernandez, D.R.; Doherty, E.; Qian, Y.; Vas, G.; Niland, B.; Telarico, T.; Garba, A.; Banerjee, S.; Middleton, F.A.; et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J. Clin. Investig. 2009, 119, 1546–1557. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Benhamed, F.; Filhoulaud, G.; Caron, S.; Lefebvre, P.; Staels, B.; Postic, C. O-GlcNAcylation Links ChREBP and FXR to Glucose-Sensing. Front. Endocrinol. 2015, 5, 230. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferré, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E358–E364. [Google Scholar] [CrossRef]
- Niwa, H.; Iizuka, K.; Kato, T.; Wu, W.; Tsuchida, H.; Takao, K.; Horikawa, Y.; Takeda, J. ChREBP Rather Than SHP Regulates Hepatic VLDL Secretion. Nutrients 2018, 10, 321. [Google Scholar] [CrossRef]
- Poungvarin, N.; Chang, B.; Imamura, M.; Chen, J.; Moolsuwan, K.; Sae-Lee, C.; Li, W.; Chan, L. Genome-Wide Analysis of ChREBP Binding Sites on Male Mouse Liver and White Adipose Chromatin. Endocrinology 2015, 156, 1982–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.B.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 6, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribback, S.; Sonke, J.; Lohr, A.; Frohme, J.; Peters, K.; Holm, J.; Peters, M.; Cigliano, A.; Calvisi, D.F.; Dombrowski, F. Hepatocellular glycogenotic foci after combined intraportal pancreatic islet transplantation and knockout of the carbohydrate responsive element binding protein in diabetic mice. Oncotarget 2017, 8, 104315–104329. [Google Scholar] [CrossRef] [PubMed]
- Ribback, S.; Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Pes, G.M.; Porcu, A.; Pascale, R.M.; Li, L.; Qiao, Y.; et al. Oncogene-dependent addiction to carbohydrate-responsive element binding protein in hepatocellular carcinoma. Cell Cycle 2018, 17, 1496–1512. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Wahed, A.; Gautier-Stein, A.; Casteras, S.; Soty, M.; Roussel, D.; Romestaing, C.; Guillou, H.; Tourette, J.A.; Pleche, N.; Zitoun, C.; et al. A link between hepatic glucose production and peripheral energy metabolism via hepatokines. Mol. Metab. 2014, 3, 531–543. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Calderaro, J.; Monteillet, L.; Silva, M.; Raffin, M.; Brevet, M.; Romestaing, C.; Roussel, D.; Zucman-Rossi, J.; Mithieux, G.; et al. Dietary exacerbation of metabolic stress leads to accelerated hepatic carcinogenesis in glycogen storage disease type Ia. J. Hepatol. 2018, 69, 1074–1087. [Google Scholar] [CrossRef]
- Eissing, L.; Scherer, T.; Tödter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef] [Green Version]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFerrin, L.G.; Atchley, W.R. A novel N-terminal domain may dictate the glucose response of Mondo proteins. PLoS ONE 2012, 7, e34803. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [Green Version]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation Increases ChREBP Protein Content and Transcriptional Activity in the Liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Glucose activates ChREBP by increasing its rate of nuclear entry and relieving repression of its transcriptional activity. J. Biol. Chem. 2008, 283, 24029–24038. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite Regulation of Nuclear Localization of Carbohydrate-response Element-binding Protein (ChREBP): ROLE OF AMP AS AN ALLOSTERIC INHIBITOR. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [Green Version]
- Unnikrishnan, R.; Pradeepa, R.; Joshi, S.R.; Mohan, V. Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 2017, 66, 1432–1442. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus--present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Accili, D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011, 14, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.W.; Adams, L.A. Non-alcoholic fatty liver disease. Crit. Rev. Clin. Lab. Sci. 2011, 48, 97–113. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metab. Clin. Exp. 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [Green Version]
- Williamson, R.M.; Price, J.F.; Glancy, S.; Perry, E.; Nee, L.D.; Hayes, P.C.; Frier, B.M.; Van Look, L.A.F.; Johnston, G.I.; Reynolds, R.M.; et al. Prevalence of and risk factors for hepatic steatosis and nonalcoholic Fatty liver disease in people with type 2 diabetes: The Edinburgh Type 2 Diabetes Study. Diabetes Care 2011, 34, 1139–1144. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaka, T.; Shimano, H. Insulin-dependent and -independent regulation of sterol regulatory element-binding protein-1c. J. Diabetes Investig. 2013, 4, 411–412. [Google Scholar] [CrossRef] [Green Version]
- Grefhorst, A.; Schreurs, M.; Oosterveer, M.H.; Cortés, V.A.; Havinga, R.; Herling, A.W.; Reijngoud, D.J.; Groen, A.K.; Kuipers, F. Carbohydrate-response-element-binding protein (ChREBP) and not the liver X receptor α (LXRα) mediates elevated hepatic lipogenic gene expression in a mouse model of glycogen storage disease type 1. Biochem. J. 2010, 432, 249–254. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Bandsma, R.H.J.; Prinsen, B.H.; de Sain-van der Velden, M.; Rake, J.P.; Boer, T.; Smit, G.P.A.; Reijngoud, D.J.; Kuipers, F. Increased de novo Lipogenesis and Delayed Conversion of Large VLDL into Intermediate Density Lipoprotein Particles Contribute to Hyperlipidemia in Glycogen Storage Disease Type 1a. Pediatr. Res. 2008, 63, 702–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjorgjieva, M.; Oosterveer, M.H.; Mithieux, G.; Rajas, F. Mechanisms by Which Metabolic Reprogramming in GSD1 Liver Generates a Favorable Tumorigenic Environment. J. Inborn Errors Metab. Screen. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Calzadilla Bertot, L.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.; Poklepovic, A.; Moyneur, E.; Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 2010, 26, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.; Stål, P.; Wahlin, S.; Björkström, N.K.; Hagström, H. Characteristics and outcome of hepatocellular carcinoma in patients with NAFLD without cirrhosis. Liver Int. 2019, 39, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Monteillet, L.; Gjorgjieva, M.; Silva, M.; Verzieux, V.; Imikirene, L.; Duchampt, A.; Guillou, H.; Mithieux, G.; Rajas, F. Intracellular lipids are an independent cause of liver injury and chronic kidney disease in non alcoholic fatty liver disease-like context. Mol. Metab. 2018, 16, 100–115. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Waskowicz, L.R.; Zhou, J.; Landau, D.J.; Brooks, E.D.; Lim, A.; Yavarow, Z.A.; Kudo, T.; Zhang, H.; Wu, Y.; Grant, S.; et al. Bezafibrate induces autophagy and improves hepatic lipid metabolism in glycogen storage disease type Ia. Hum. Mol. Genet. 2018, 28, 143–154. [Google Scholar] [CrossRef]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Müller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell 2016, 167, 843–857. [Google Scholar] [CrossRef]
- Perry, R.J.; Zhang, D.; Zhang, X.M.; Boyer, J.L.; Shulman, G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 2015, 347, 1253–1256. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Kim, T.; Zhang, X.M.; Lee, H.Y.; Pesta, D.; Popov, V.B.; Zhang, D.; Rahimi, Y.; Jurczak, M.J.; Cline, G.W.; et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013, 18, 740–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajas, F.; Gautier-Stein, A.; Mithieux, G. Glucose-6 Phosphate, a Central Hub for Liver Carbohydrate Metabolism. Metabolites 2019, 9, 282. https://doi.org/10.3390/metabo9120282
Rajas F, Gautier-Stein A, Mithieux G. Glucose-6 Phosphate, a Central Hub for Liver Carbohydrate Metabolism. Metabolites. 2019; 9(12):282. https://doi.org/10.3390/metabo9120282
Chicago/Turabian StyleRajas, Fabienne, Amandine Gautier-Stein, and Gilles Mithieux. 2019. "Glucose-6 Phosphate, a Central Hub for Liver Carbohydrate Metabolism" Metabolites 9, no. 12: 282. https://doi.org/10.3390/metabo9120282
APA StyleRajas, F., Gautier-Stein, A., & Mithieux, G. (2019). Glucose-6 Phosphate, a Central Hub for Liver Carbohydrate Metabolism. Metabolites, 9(12), 282. https://doi.org/10.3390/metabo9120282