Microbial Transformations of Organically Fermented Foods


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Sample Collection
2.2. Extractions
2.3. Mass Spectrometry
2.4. Metabolome Data Analysis
2.5. Microbiome Sequencing
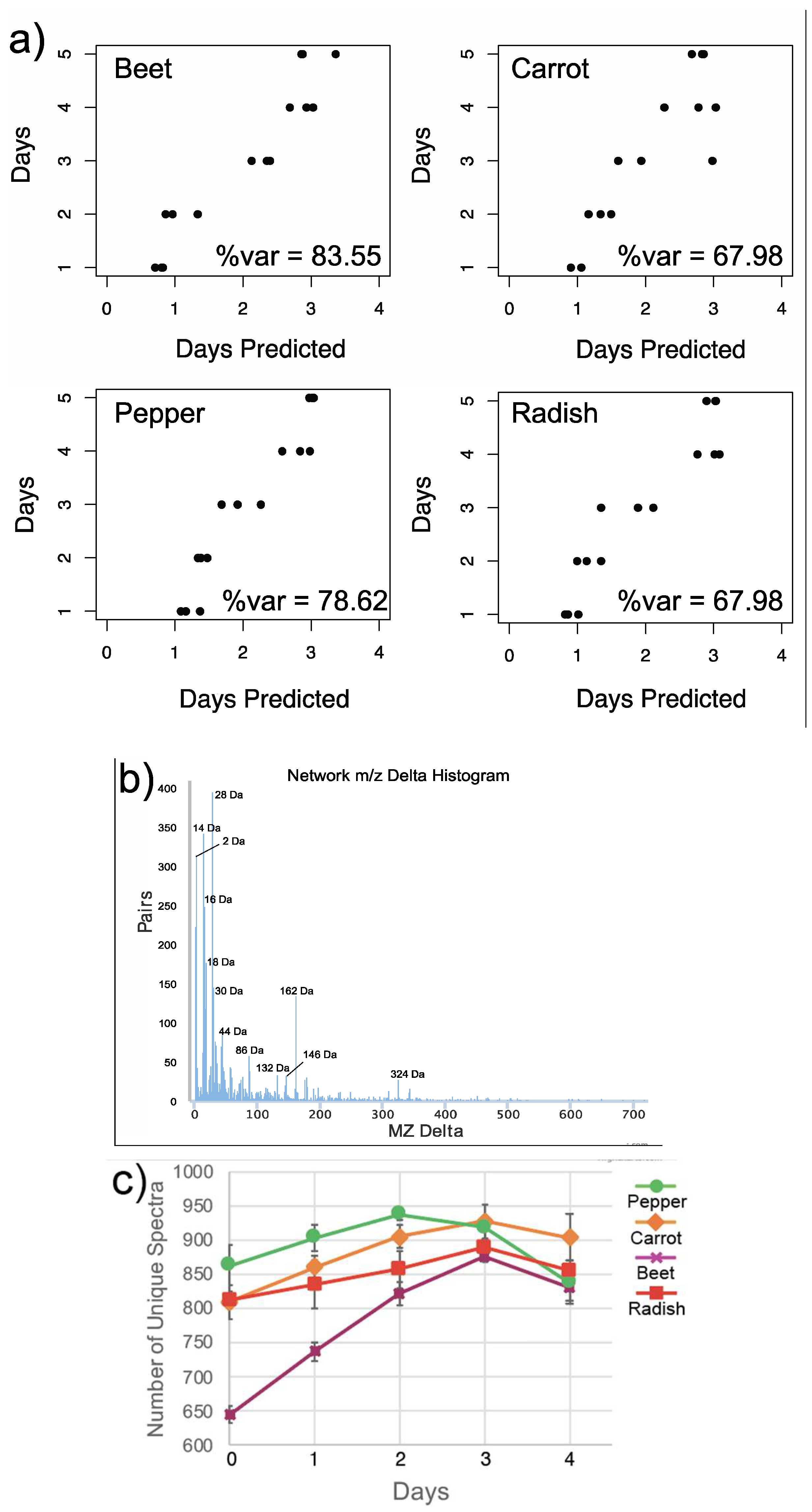
2.6. Statistical Analysis
3. Results
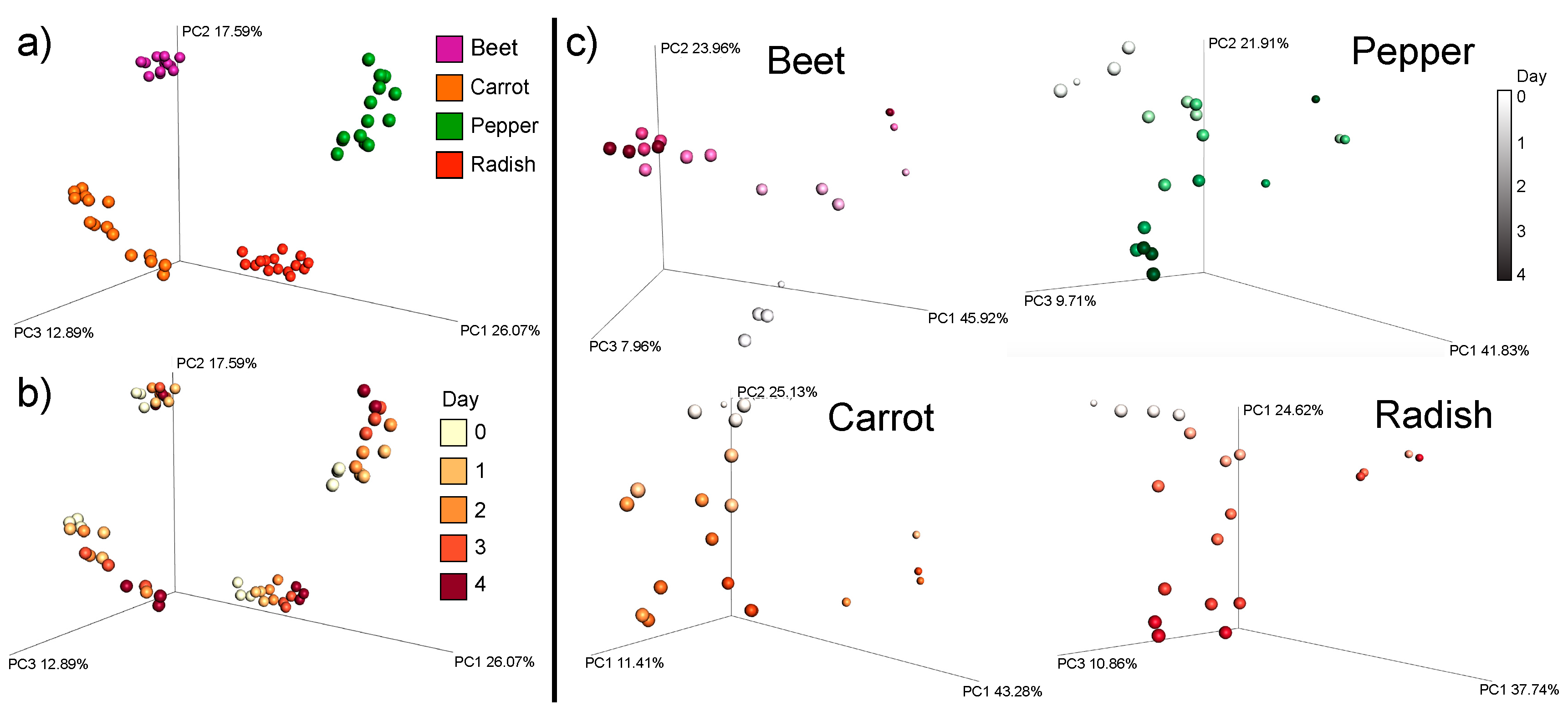
3.1. Overall Metabolome Changes in Organic Fermented Foods
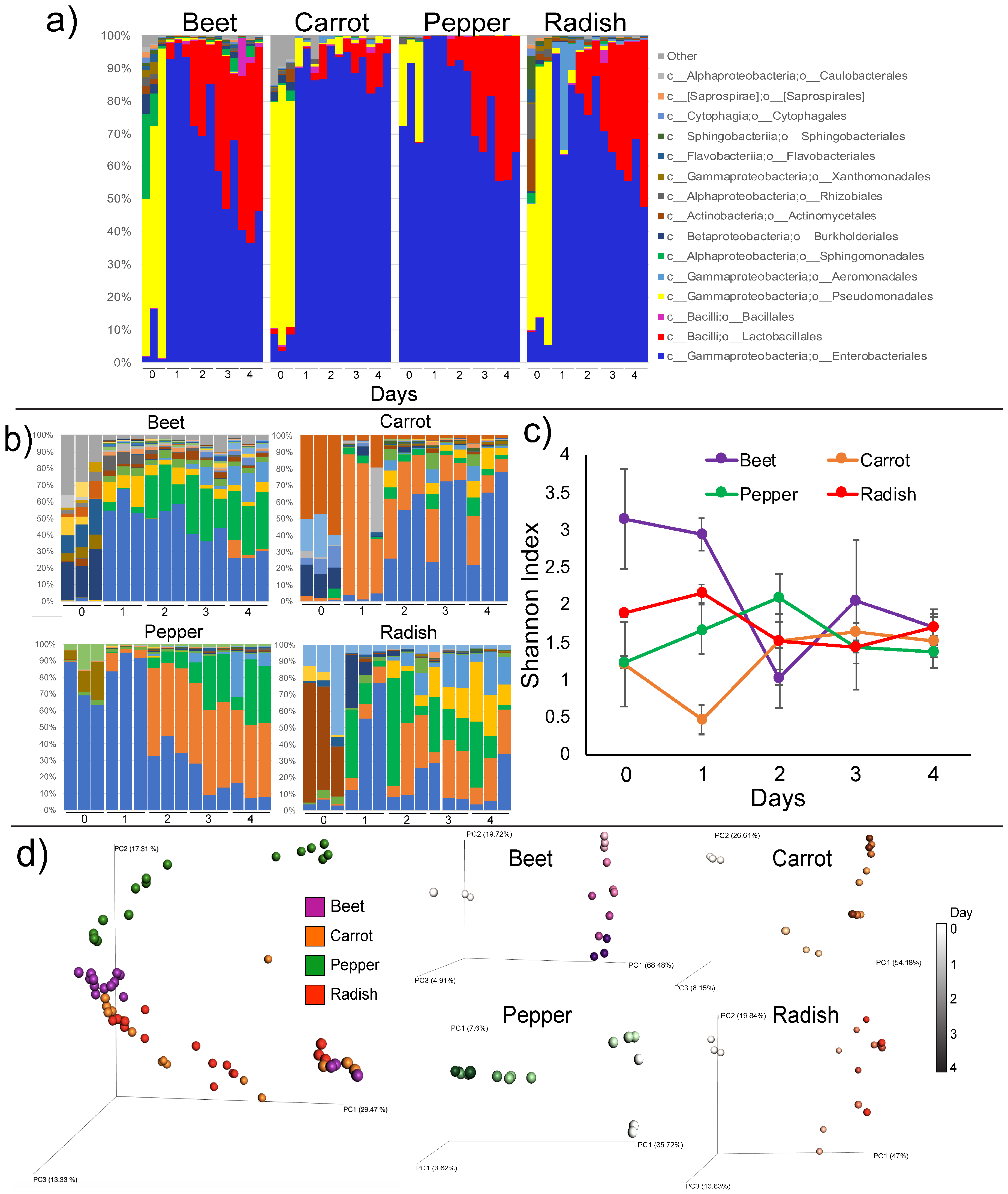
3.2. Microbiome Dynamics
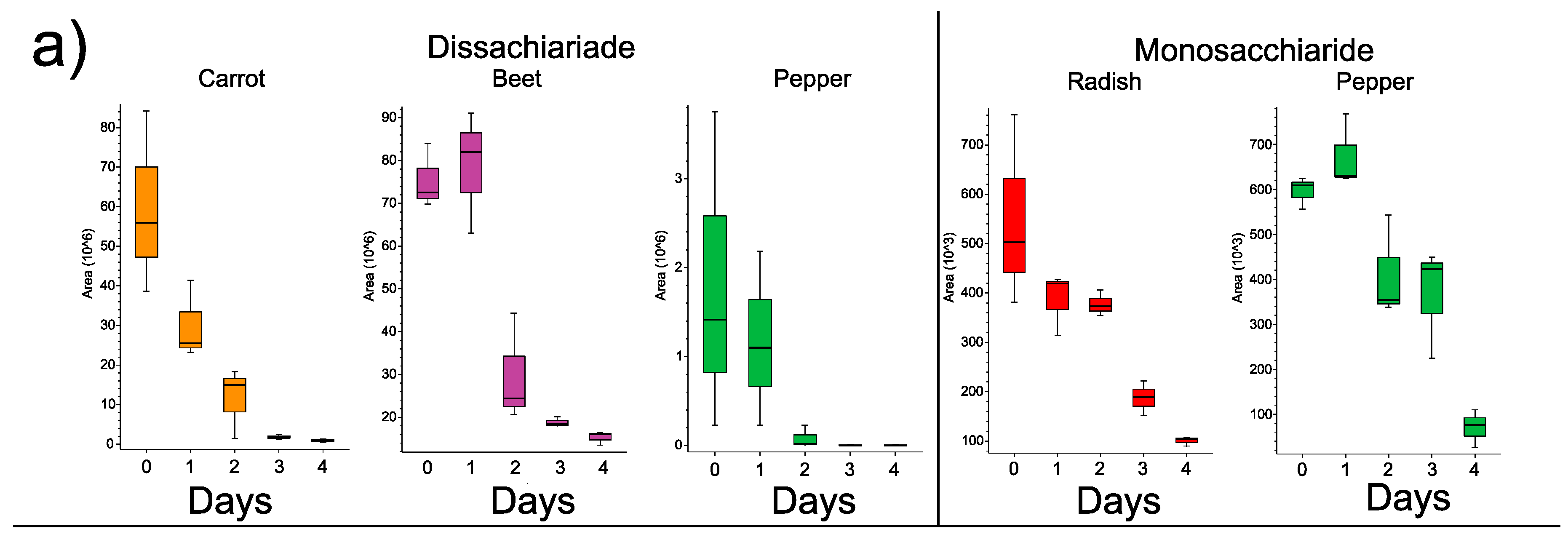
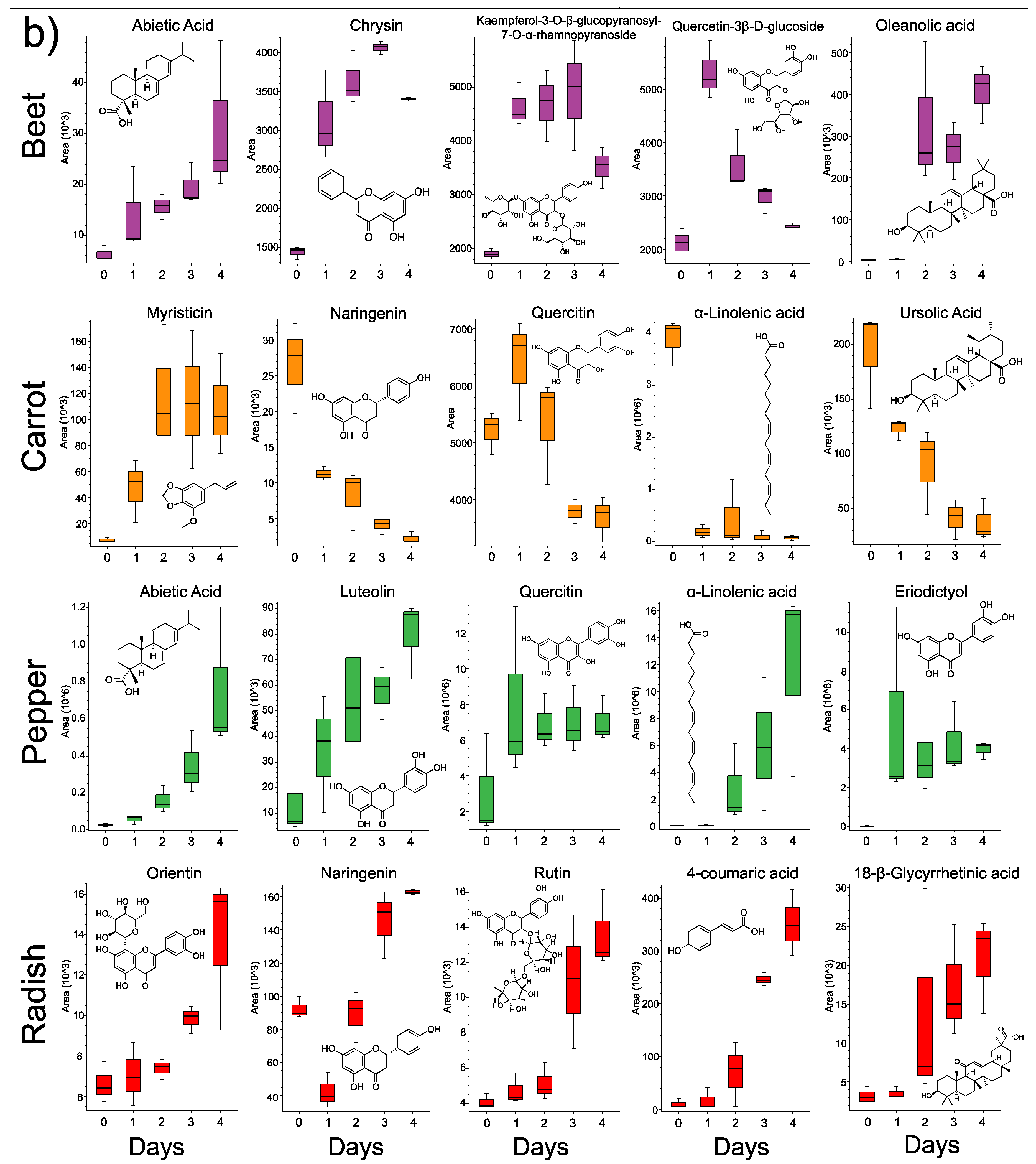
3.3. Changes in Individual Metabolites through Time
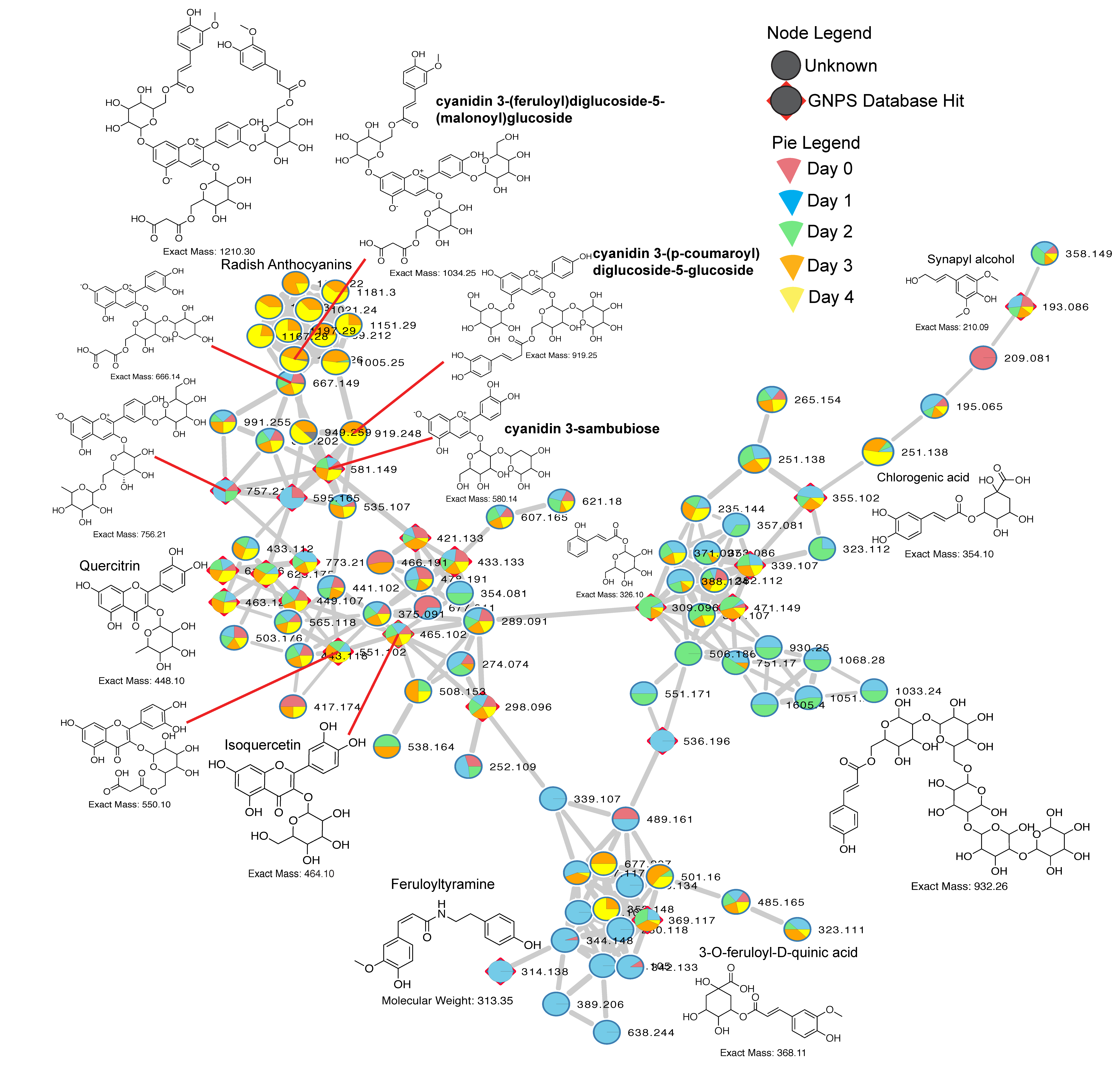
3.4. Anthocyanin Dynamics in Radishes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell-Platt, G. Fermented foods—A world perspective. Food Res. Int. 1994, 27, 253–257. [Google Scholar] [CrossRef]
- Selhub, E.M.; Logan, A.C.; Bested, A.C. Fermented foods, microbiota, and mental health: Ancient practice meets nutritional psychiatry. J. Physiol. Anthropol. 2014, 33, 2. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.E.; Dutton, R.J. Fermented foods as experimentally tractable microbial ecosystems. Cell 2015, 161, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.; Embriette, H.; Justine W., D.; James T., M.; Antonio, G.; Gail, A.; Alexander A., A.; Bahar, B.; Caitriona, B.; Yingfeng, C.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2013, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.; Knight, R.; Blakeslee, S. Dirt is Good: The Advantage of Germs for Your Child’s Developing Immune System; St. Martin’s Press: New York, NY, USA, 2017. [Google Scholar]
- Park, S.E.; Yoo, S.A.; Seo, S.H.; Lee, K.I.; Na, C.S.; Son, H.S. GC–MS based metabolomics approach of Kimchi for the understanding of Lactobacillus plantarum fermentation characteristics. LWT—Food Sci. Technol. 2016, 68, 313–321. [Google Scholar] [CrossRef]
- Jung, J.Y.; Lee, S.H.; Kim, J.M.; Park, M.S.; Bae, J.W.; Hahn, Y.; Madsen, E.L.; Jeon, C.O. Metagenomic analysis of kimchi, a traditional Korean fermented food. Appl. Environ. Microbiol. 2011, 77, 2264–2274. [Google Scholar] [CrossRef]
- Mugula, J.; Nnko, S.A.; Narvhus, J.; Sørhaug, T. Microbiological and fermentation characteristics of togwa, a Tanzanian fermented food. Int. J. Food Microbiol. 2003, 80, 187–199. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vázquez-Baeza, Y.; Ackermann, G.; DeReus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, web-enabled microbiome meta-analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, 00191-16. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.C.; Petras, D.; Quinn, R.A.; Protsyuk, I.; Archer, F.I.; Ransome, E.; Williams, G.J.; Bailey, B.A.; Vermeij, M.J.A.; Alexandrov, T.; et al. Meta-mass shift chemical profiling of metabolomes from coral reefs. Proc. Natl. Acad. Sci. USA 2017, 114, 11685–11690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Prior, R.L. Identification and Characterization of Anthocyanins by High-Performance Liquid Chromatography−Electrospray Ionization−Tandem Mass Spectrometry in Common Foods in the United States: Vegetables, Nuts, and Grains. J. Agric. Food Chem. 2005, 53, 3101–3113. [Google Scholar] [CrossRef]
- Rezac, S.; Kok, C.R.; Heermann, M.; Hutkins, R. Fermented Foods as a Dietary Source of Live Organisms. Front. Microbiol. 2018, 9, 1785. [Google Scholar] [CrossRef]
- Chilton, S.N.; Burton, J.P.; Reid, G. Inclusion of Fermented Foods in Food Guides around the World. Nutrients 2015, 7, 390. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; Voort, M.v.d.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, 1743–1752. [Google Scholar] [CrossRef]
- Wang, M.; J Carver, J.; V Phelan, V.; M Sanchez, L.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; A Kapono, C.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Perez Diaz, I.M.; Breidt, F.; Buescher, R.W.; Arroyo-Lopez, F.N.; Jimenez-Diaz, R.; Bautista-Gallego, J.; Garrido-Fernandez, A.; Yoon, S.; Johanningsmeier, S.D. Fermented and Acidified Vegetables. In Compendium of Methods for the Microbiological Examination of Foods; American Public Health Association: Washington, DC, USA, 2015; Volume 51, ISBN 978-0-87553-022-2. [Google Scholar]
- Fang, F.; Xu, J.; Li, Q.; Xia, X.; Du, G. Characterization of a Lactobacillus brevis strain with potential oral probiotic properties. BMC Microbiol. 2018, 18, 221. [Google Scholar] [CrossRef]
- Wuyts, S.; Beeck, W.V.; Oerlemans, E.F.M.; Wittouck, S.; Claes, I.J.J.; Boeck, I.D.; Weckx, S.; Lievens, B.; Vuyst, L.D.; Lebeer, S. Carrot Juice Fermentations as Man-Made Microbial Ecosystems Dominated by Lactic Acid Bacteria. Appl. Environ. Microbiol. 2018, 84, e00134-18. [Google Scholar] [CrossRef] [Green Version]
- Zabat, M.; Sano, W.; Wurster, J.; Cabral, D.; Belenky, P. Microbial Community Analysis of Sauerkraut Fermentation Reveals a Stable and Rapidly Established Community. Foods 2018, 7, 77. [Google Scholar] [CrossRef]
- Tolan, J.S.; Finn, R.K. Fermentation of d-Xylose and l-Arabinose to Ethanol by Erwinia chrysanthemi. Appl. Environ. Microbiol. 1987, 53, 2033–2038. [Google Scholar]
- Tamang, J.P.; Watanabe, K.; Holzapfel, W.H. Review: Diversity of Microorganisms in Global Fermented Foods and Beverages. Front Microbiol. 2016, 7, 377. [Google Scholar] [CrossRef] [Green Version]
- Leff, J.W.; Fierer, N. Bacterial Communities Associated with the Surfaces of Fresh Fruits and Vegetables. PLoS ONE 2013, 8, e59310. [Google Scholar] [CrossRef]
- Leroy, F.; De Vuyst, L. Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci. Technol. 2004, 15, 67–78. [Google Scholar] [CrossRef]
- Jiang, N.; Doseff, A.; Grotewold, E. Flavones: From Biosynthesis to Health Benefits. Plants 2016, 5, 27. [Google Scholar] [CrossRef]
- Yao, L.H.; Jiang, Y.M.; Shi, J.; Tomas-Barberan, F.A.; Datta, N.; Singanusong, R.; Chen, S.S. Flavonoids in food and their health benefits. Plant. Foods Hum. Nutr. 2004, 59, 113–122. [Google Scholar] [CrossRef]
- Pojer, E.; Mattivi, F.; Johnson, D.; Stockley, C.S. The Case for Anthocyanin Consumption to Promote Human Health: A Review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 483–508. [Google Scholar] [CrossRef]
- Shi, J.; Arunasalam, K.; Yeung, D.; Kakuda, Y.; Mittal, G.; Jiang, Y. Saponins from Edible Legumes: Chemistry, Processing, and Health Benefits. J. Med. Food 2004, 7, 67–78. [Google Scholar] [CrossRef]
- Justesen, U.; Arrigoni, E.; Larsen, B.R.; Amado, R. Degradation of Flavonoid Glycosides and Aglycones During in vitro Fermentation with Human Faecal Flora. LWT—Food Sci. Technol. 2000, 33, 424–430. [Google Scholar] [CrossRef]
- Hostetler, G.; Riedl, K.; Cardenas, H.; Diosa-Toro, M.; Arango, D.; Schwartz, S.; Doseff, A.I. Flavone deglycosylation increases their anti-inflammatory activity and absorption. Mol. Nutr. Food Res. 2012, 56, 558–569. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raghuvanshi, R.; Grayson, A.G.; Schena, I.; Amanze, O.; Suwintono, K.; Quinn, R.A. Microbial Transformations of Organically Fermented Foods. Metabolites 2019, 9, 165. https://doi.org/10.3390/metabo9080165
Raghuvanshi R, Grayson AG, Schena I, Amanze O, Suwintono K, Quinn RA. Microbial Transformations of Organically Fermented Foods. Metabolites. 2019; 9(8):165. https://doi.org/10.3390/metabo9080165
Chicago/Turabian StyleRaghuvanshi, Ruma, Allyssa G. Grayson, Isabella Schena, Onyebuchi Amanze, Kezia Suwintono, and Robert A. Quinn. 2019. "Microbial Transformations of Organically Fermented Foods" Metabolites 9, no. 8: 165. https://doi.org/10.3390/metabo9080165
APA StyleRaghuvanshi, R., Grayson, A. G., Schena, I., Amanze, O., Suwintono, K., & Quinn, R. A. (2019). Microbial Transformations of Organically Fermented Foods. Metabolites, 9(8), 165. https://doi.org/10.3390/metabo9080165