Structure Modeling of the Norepinephrine Transporter


Abstract
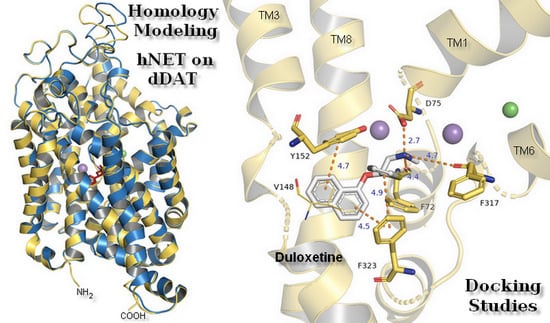
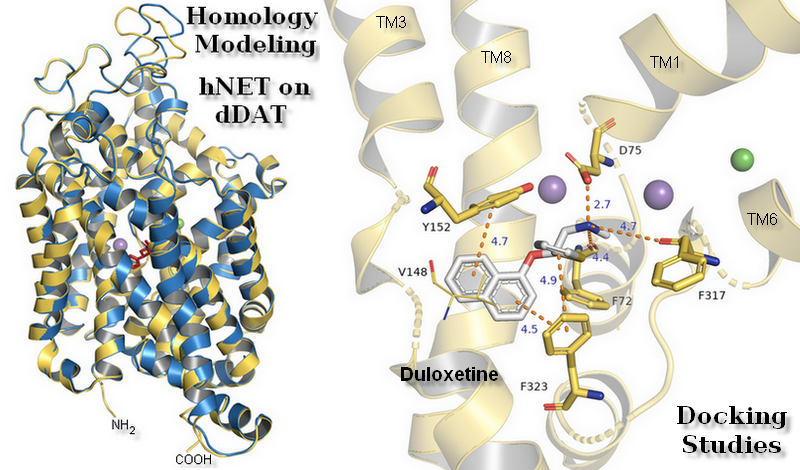
:
1. Introduction
2. Results and Discussion
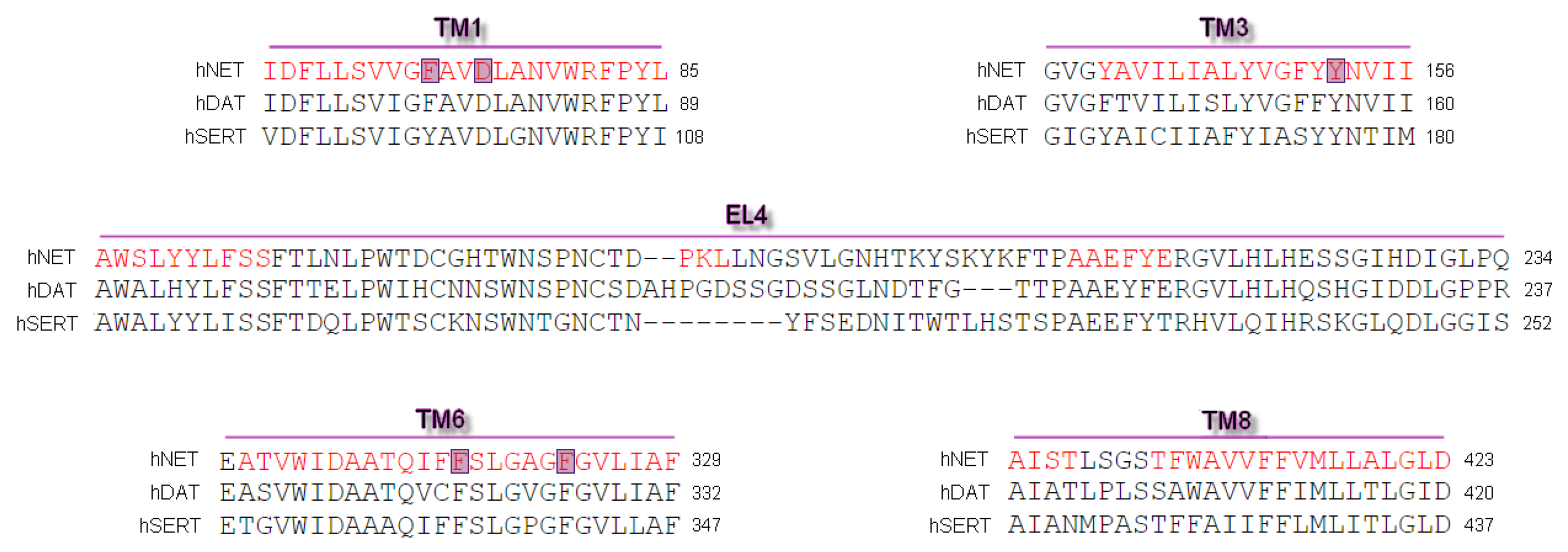
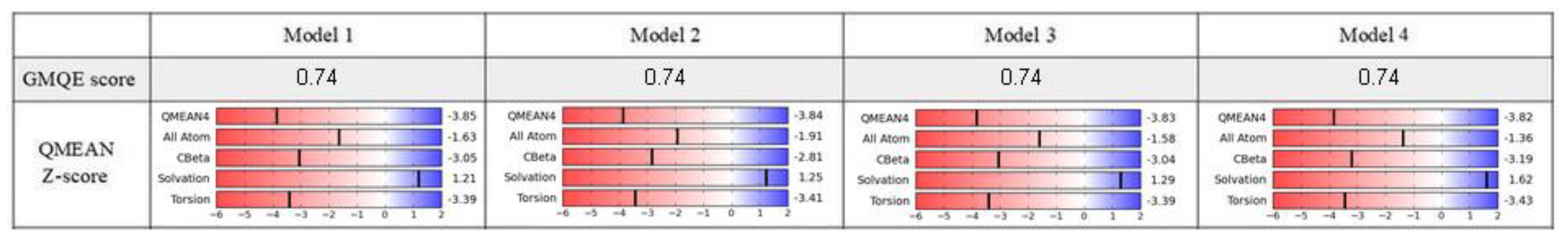
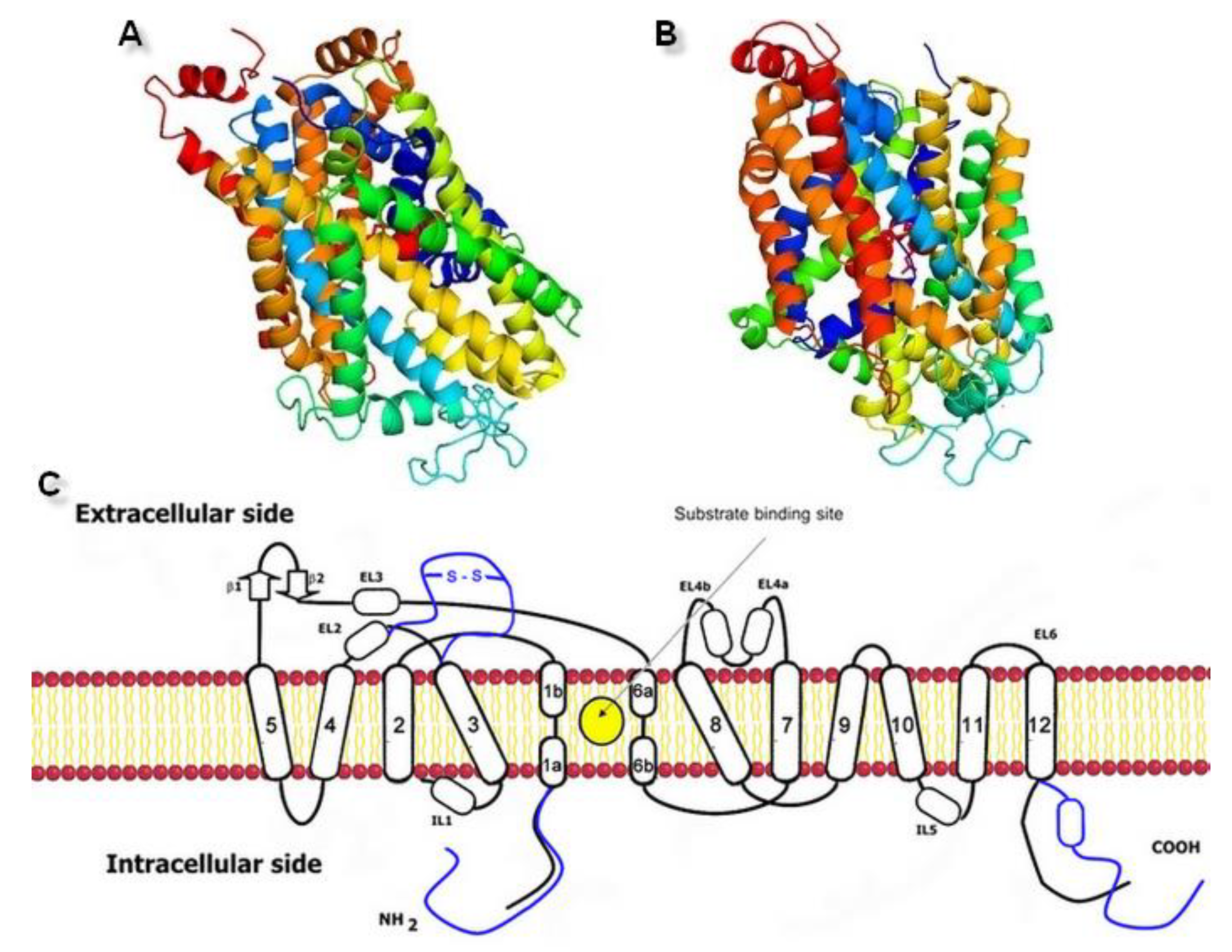
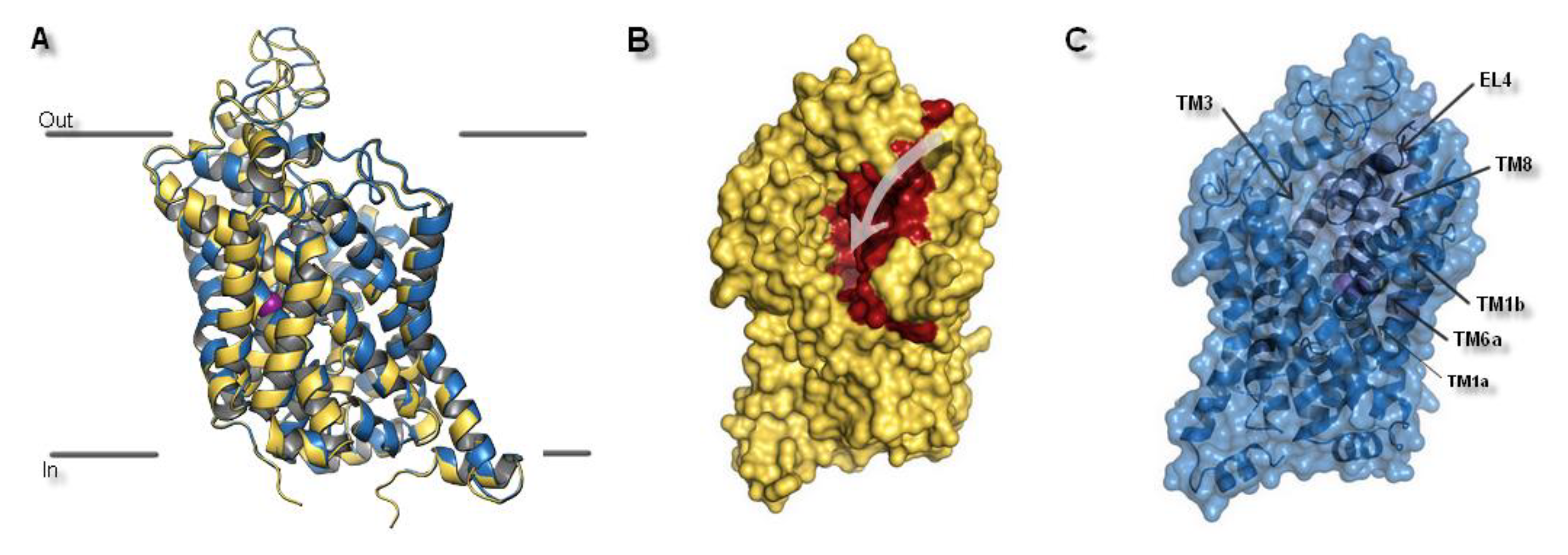
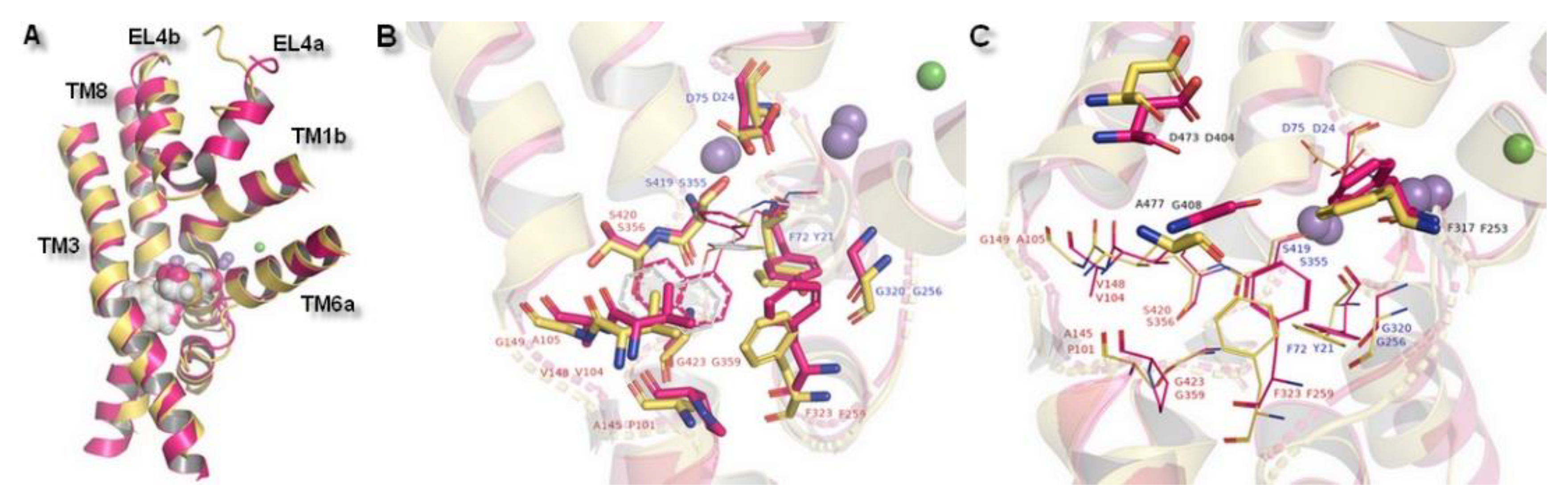
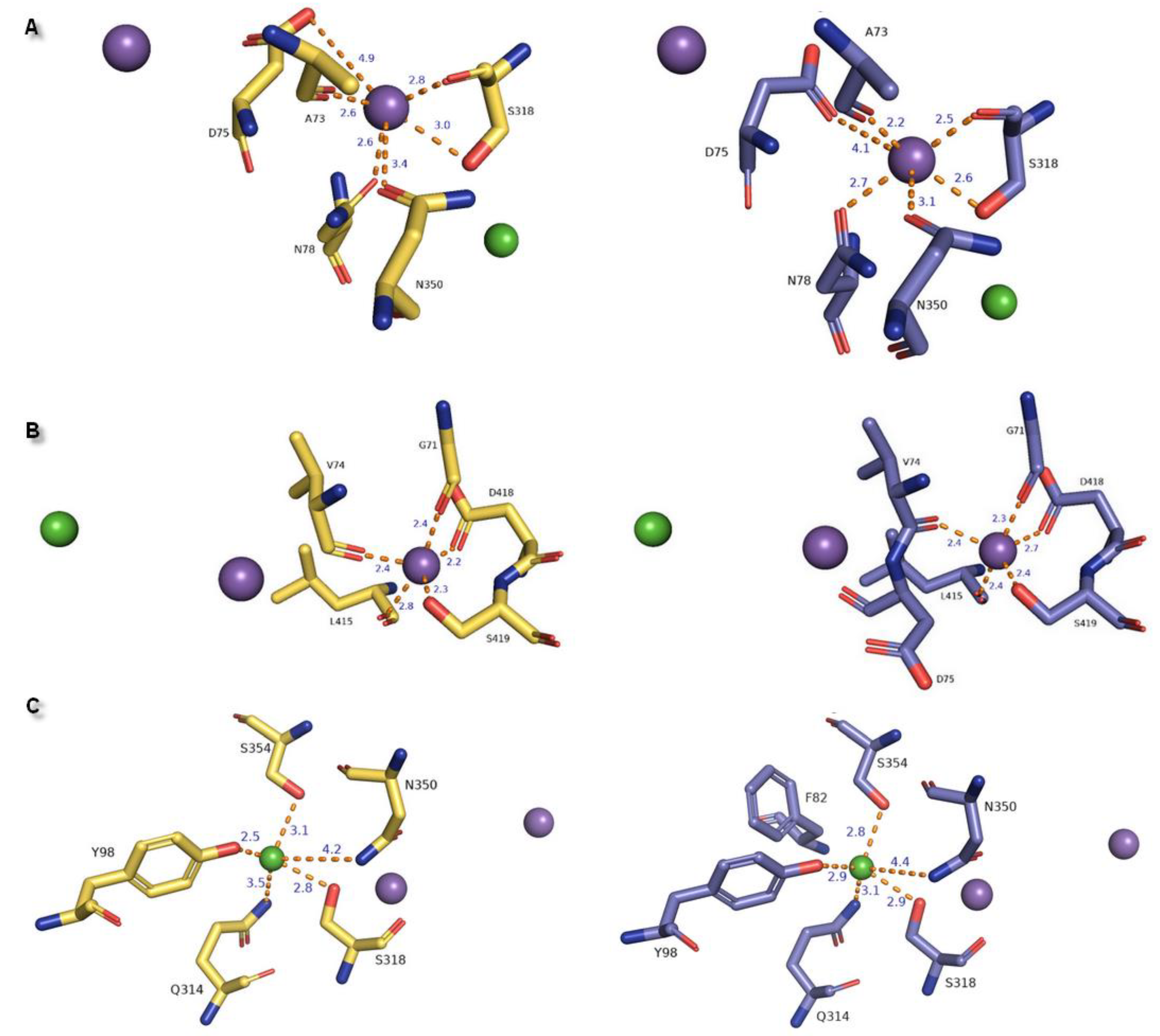
2.1. Model Building and Evaluation
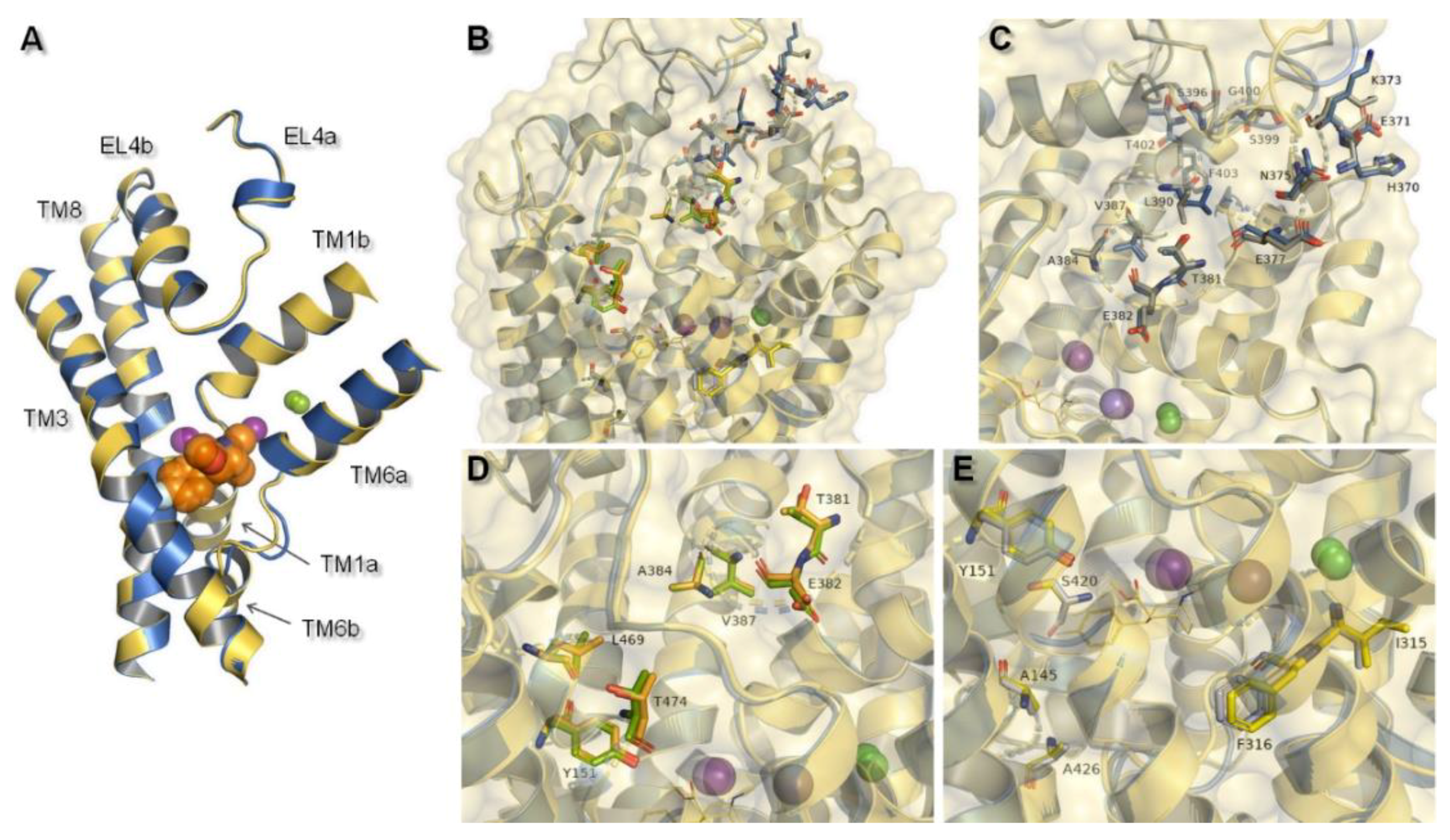
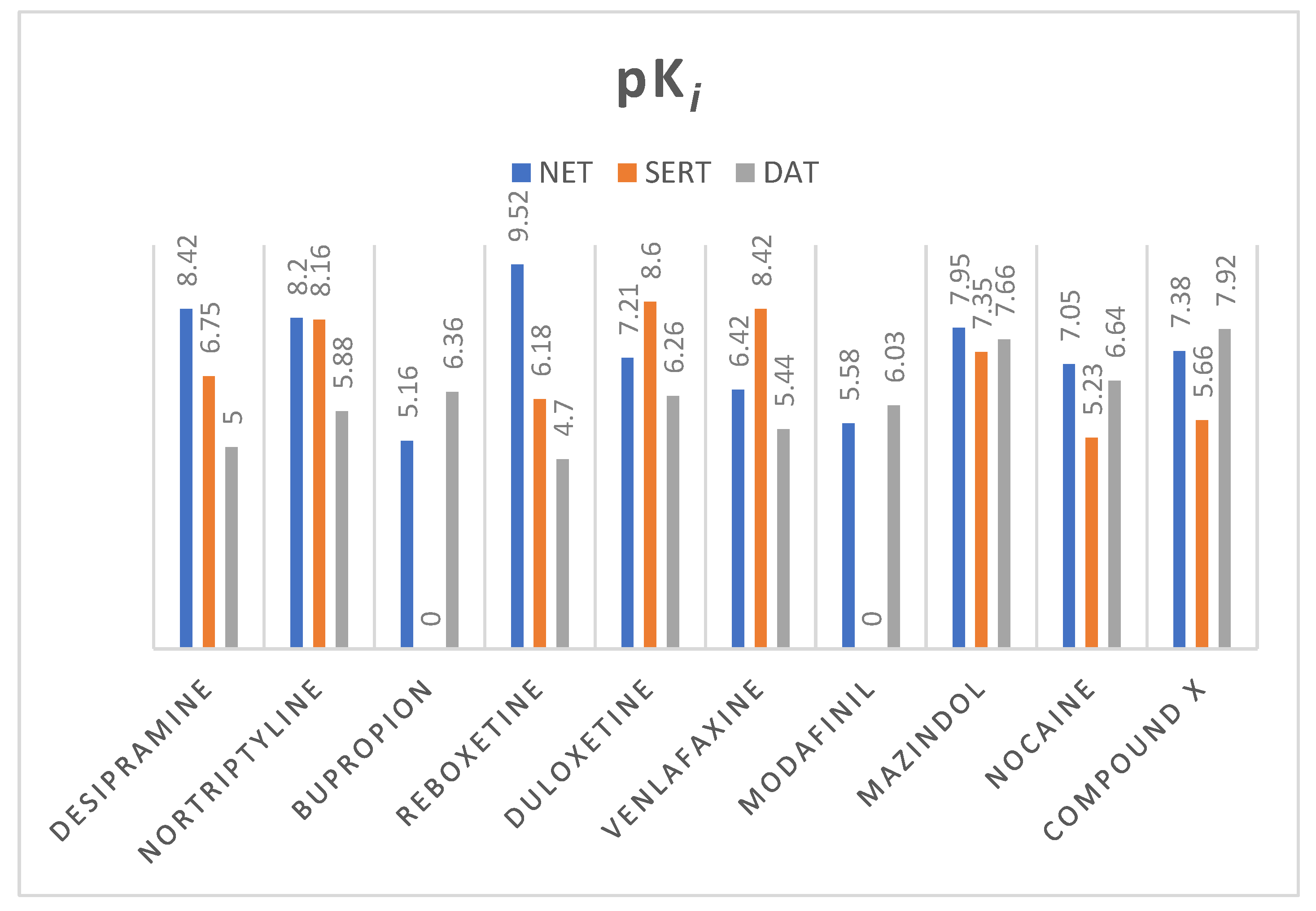
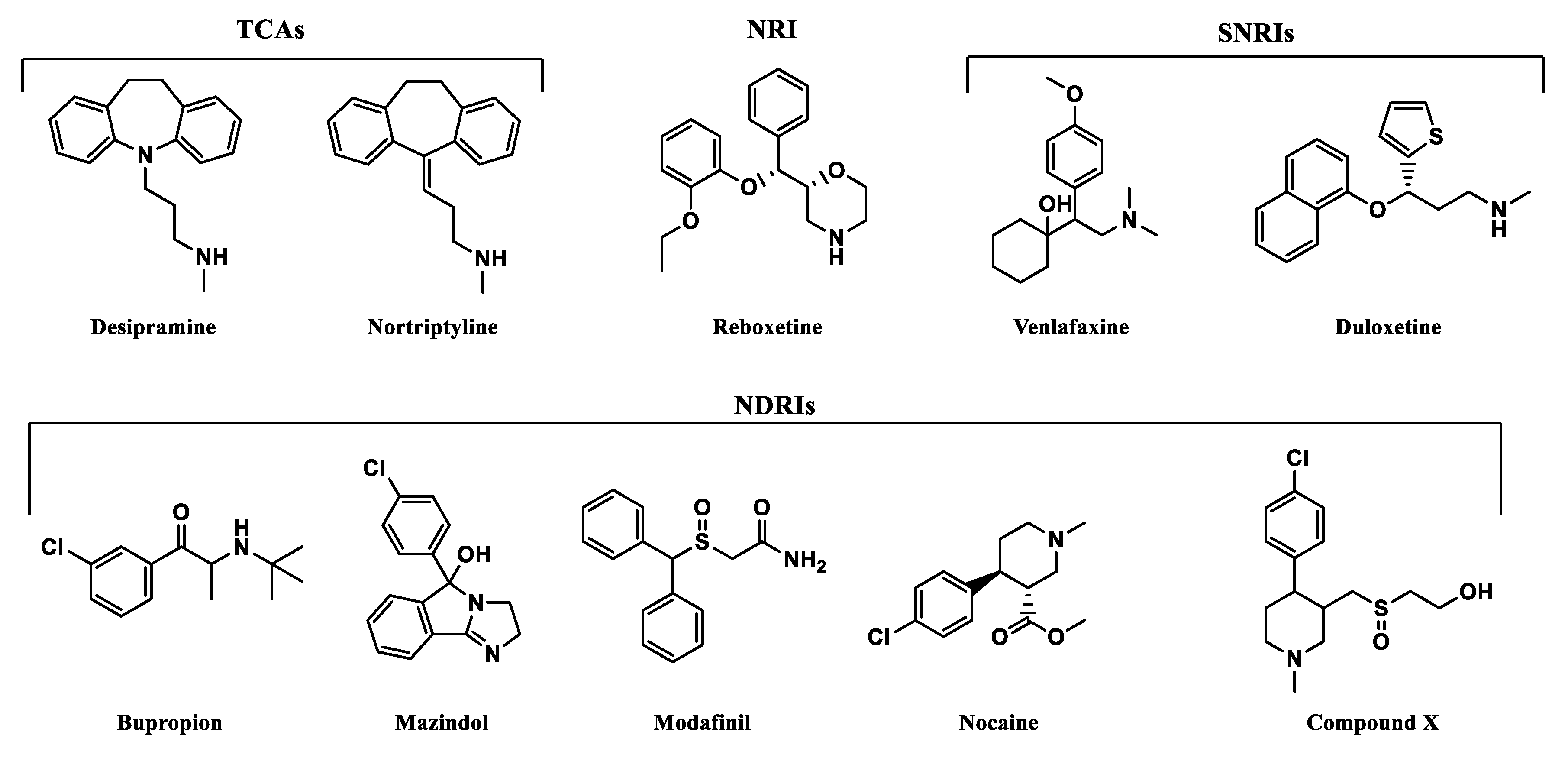
2.2. Norepinephrine Transporter Inhibitors
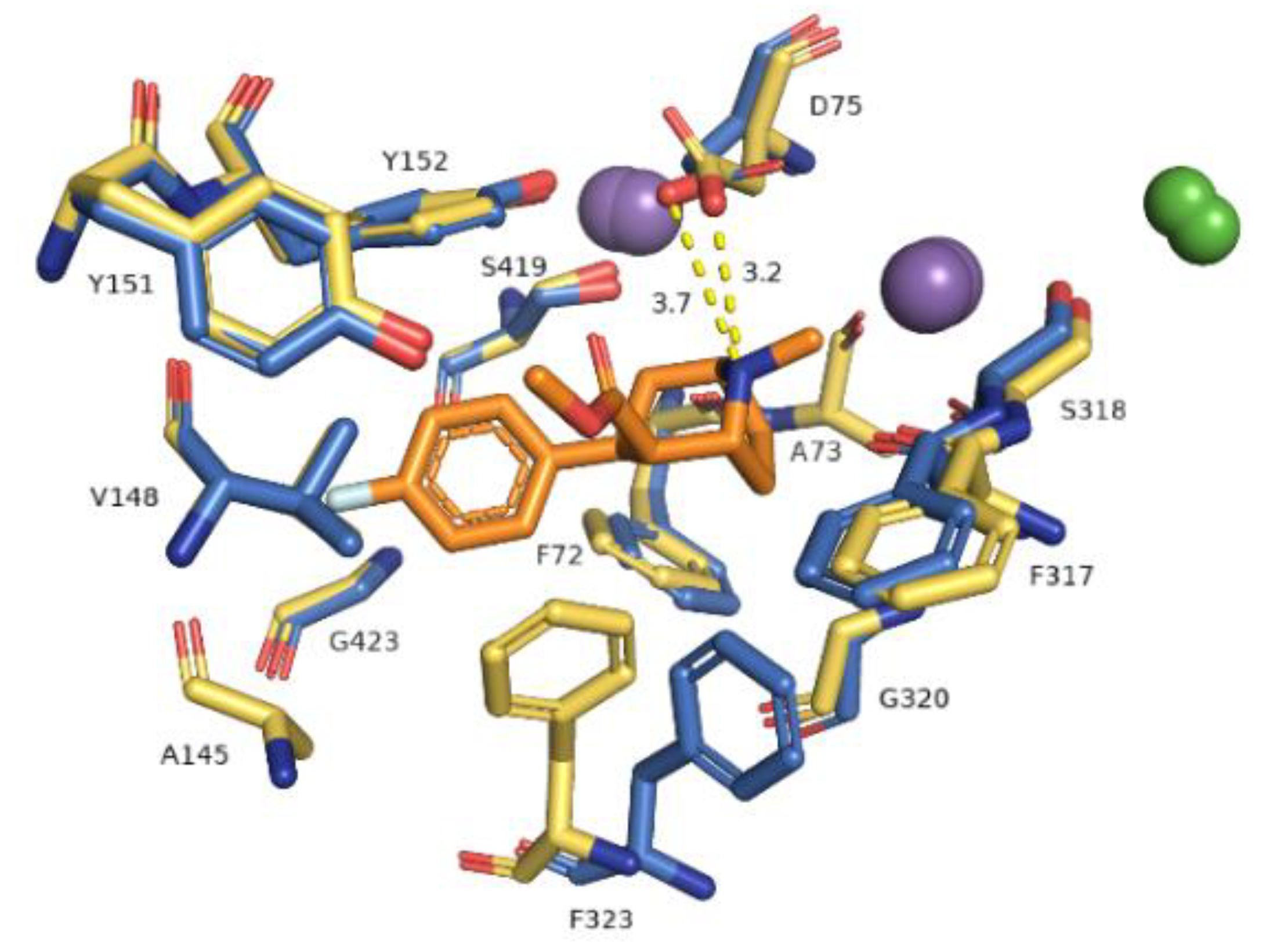
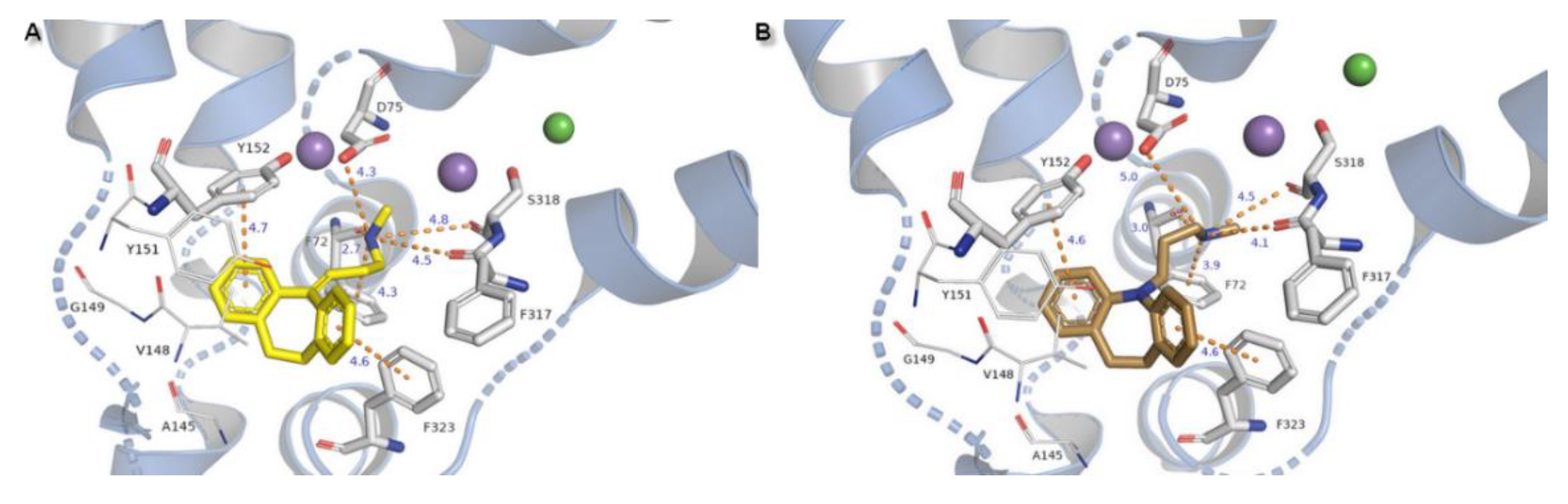
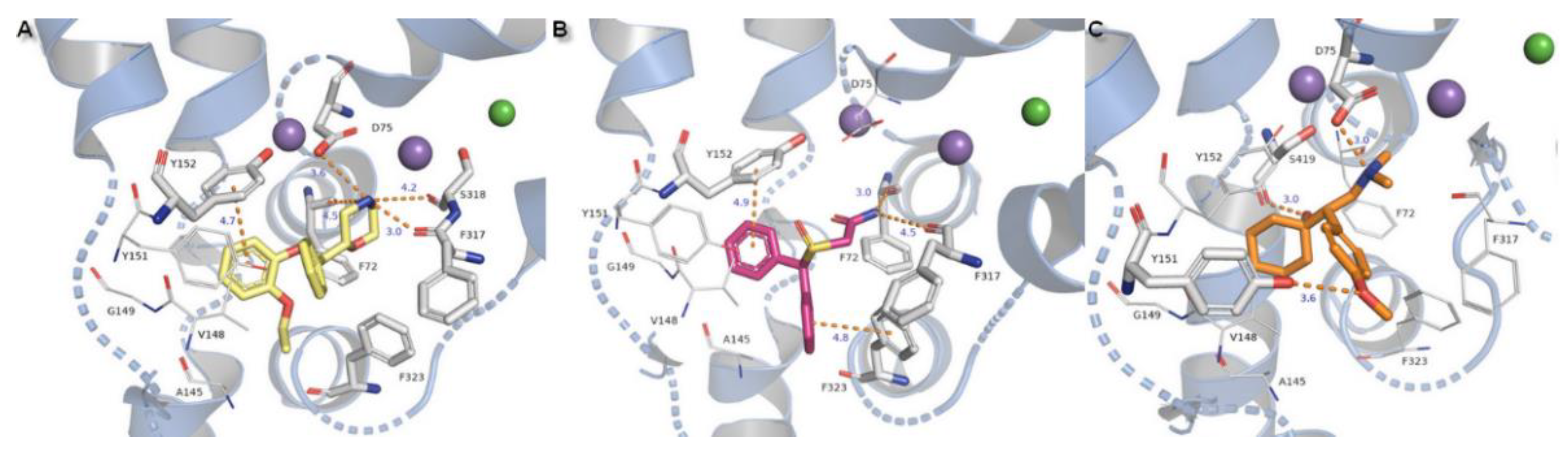
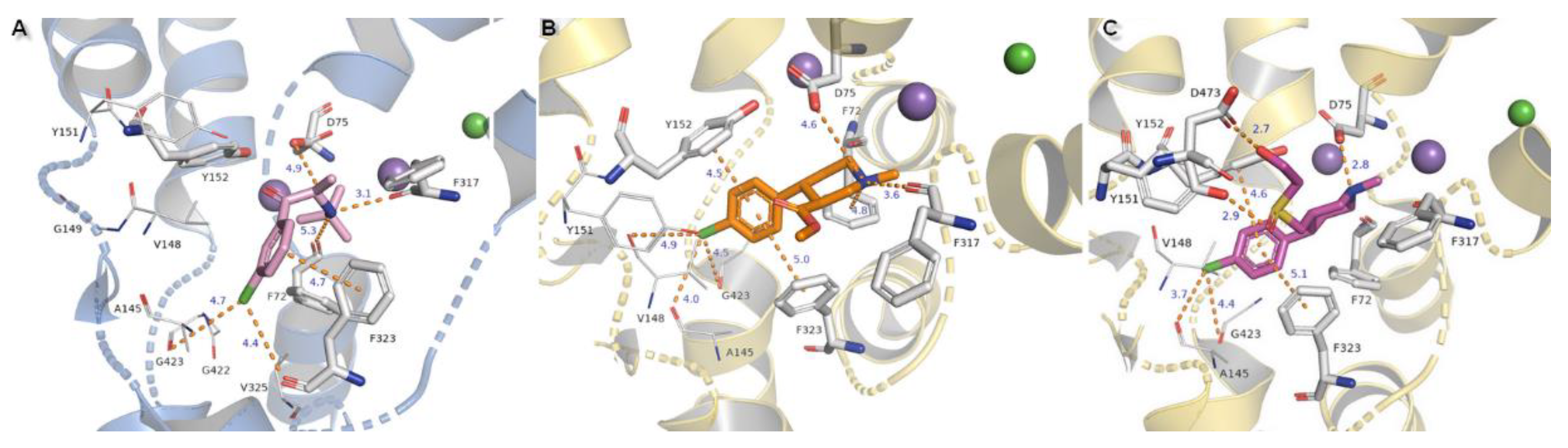
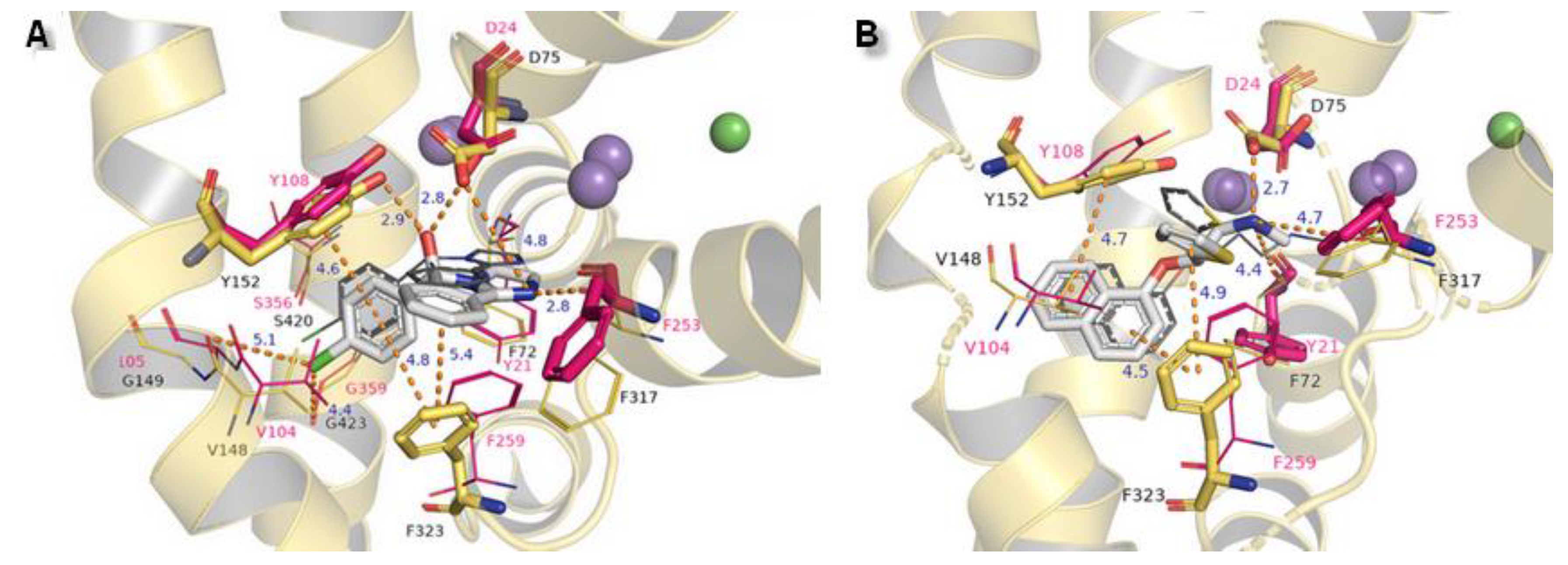
2.3. Docking Studies
3. Materials and Methods
3.1. Sequences and Homology Models
3.2. Ligands Preparation
3.3. Docking
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine, serotonin |
| ADHD | attention deficit hyperactivity disorder |
| BAT | Biogenic amine transporter |
| cDNA | Complementary DNA |
| CNS | Central nervous system |
| DAT | Dopamine transporter |
| dDAT | Drosophila dopamine transporter |
| EL2 | Extracellular loop 2 |
| EL4 | Extracellular loop 4 |
| GMQE | Global Model Quality Estimation |
| hDAT | Human dopamine transporter |
| hNET | Human norepinephrine transporter |
| IL1 | Intracellular loop 1 |
| LeuBAT | Leucine biogenic amine transporter |
| LeuT | Leucine transporter |
| MAT | Monoamine transporter |
| NDRIs | Norepinephrine/dopamine re-uptake inhibitors |
| NE | Norepinephrine |
| NET | Norepinephrine transporter |
| NRIs | Norepinephrine re-uptake inhibitors |
| NSSs | Neurotransmitter sodium symporters |
| PDB | Protein Data Bank |
| pKi | Negative logarithm of the dissociation constant of ligand—transporter complex |
| QMEAN | Qualitative Model Energy Analysis |
| SERT | Serotonin transporter |
| SLC6 | Solute carrier family 6 |
| SNRIs | Serotonin/norepinephrine re-uptake inhibitors |
| SSRIs | Selective serotonin re-uptake inhibitors |
| TCAs | Tricyclic antidepressants |
| TM | Transmembrane domain |
References
- Kanner, B.I.; Zomot, E. Sodium-Coupled Neurotransmitter Transporters. Chem. Rev. 2008, 108, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Geier, E.; Fan, H.; Irwin, J.J.; Shoichet, B.K.; Giacomini, K.M.; Sali, A. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl. Acad. Sci. USA 2011, 108, 15810–15815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandela, P.; Ordway, G.A. The norepinephrine transporter and its regulation. J. Neurochem. 2006, 97, 310–333. [Google Scholar] [CrossRef] [PubMed]
- Focke, P.J.; Wang, X.; Larsson, H.P. Neurotransmitter transporters: Structure meets function. Structure 2013, 21, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, A.S.; Andersen, J.; Jørgensen, T.N.; Sørensen, L.; Eriksen, J.; Loland, C.J.; Strømgaard, K.; Gether, U. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef]
- Zhou, J. Norepinephrine transporter inhibitors and their therapeutic potential. Drugs Future 2004, 29, 1235–1244. [Google Scholar] [CrossRef]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structure of the dopamine transporter in complex with tricyclic antidepressant. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Buck, K.J.; Amara, S.G. Chimeric dopamine-norepinephrine transporters delineate structural domains influencing selectivity for catecholamines and 1-methyl-4-phenylpyridinium. Neurobiology 1994, 91, 12584–12588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma membrane monoamine transporters: Structure, regulation and function. Nat. Rev. Neurosci. 2003, 4, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Mortensen, O. Overview of monoamine transporters. Curr. Protoc. Pharmacol. 2015, 40, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Bala, P.A.; Foster, J.; Carvelli, L.; Henry, L.K. SLC6 Transporters: Structure, Function, Regulation, Disease Association and Therapeutics. Mol. Asp. Med. 2013, 34, 197–219. [Google Scholar]
- Chen, N.H.; Reith, M.E.A.; Quick, M.W. Synaptic uptake and beyond: The sodium- and chloride-dependent neurotransmitter transporter family SLC6. Pflugers Arch. Eur. J. Physiol. 2004, 447, 519–531. [Google Scholar] [CrossRef]
- Giros, B.; Wang, Y.M.; Suter, S.; McLeskey, S.B.; Pifl, C.; Caron, M.G. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine-norepinephrine transporters. J. Biol. Chem. 1994, 269, 15985–15988. [Google Scholar]
- Beuming, T.; Shi, L.; Javitch, J.A.; Weinstein, H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006, 70, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Sucic, S.; Bryan-Lluka, L.J. Investigation of the functional roles of the MELAL and GQXXRXG motifs of the human noradrenaline transporter using cysteine mutants. Eur. J. Pharmacol. 2007, 556, 27–35. [Google Scholar] [CrossRef]
- Sucic, S.; Bryan-Lluka, L.J. Roles of transmembrane domain 2 and the first intracellular loop in human noradrenaline transporter function: Pharmacological and SCAM analysis. J. Neurochem. 2005, 94, 1620–1630. [Google Scholar] [CrossRef]
- Wenge, B.; Bönisch, H. The role of cysteines and histidins of the norepinephrine transporter. Neurochem. Res. 2013, 38, 1303–1314. [Google Scholar] [CrossRef]
- Lynagh, T.; Khamu, T.S.; Bryan-Lluka, L.J. Extracellular loop 3 of the noradrenaline transporter contributes to substrate and inhibitor selectivity. Naunyn. Schmiedebergs. Arch. Pharmacol. 2014, 387, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Loland, C.J. The use of LeuT as a model in elucidating binding sites for substrates and inhibitors in neurotransmitter transporters. Biochim. Biophys. Acta 2015, 1850, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Koldsø, H.; Christiansen, A.B.; Sinning, S.; Schiøtt, B. Comparative modeling of the human monoamine transporters: Similarities in substrate binding. ACS Chem. Neurosci. 2013, 4, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitte, H.H.; Freissmuth, M. The reverse operation of Na+/Cl−-coupled neurotransmitter transporters–why amphetamines take two to tango. J. Neurochem. 2010, 112, 340–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, H.; Gouaux, E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 2012, 481, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, L.R.; Tavoulari, S.; Zhang, Y.-W.; Rudnick, G.; Honig, B. Identification of a chloride ion binding site in Na+/Cl−–dependent transporters. Proc. Natl. Acad. Sci. USA 2007, 104, 12761–12766. [Google Scholar] [CrossRef] [Green Version]
- PROMALS3D Multiple Sequence and Structure Alignment Server. Available online: http://prodata.swmed.edu/promals3d/promals3d.php (accessed on 2 June 2019).
- Pei, J.; Grishin, N.V. AL2CO: Calculation of positional conservation in a protein sequence alignment. Bioinformatics 2001, 17, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Output of Sequence Alignment. Available online: http://prodata.swmed.edu/promals3d/info/promals_output.html (accessed on 2 June 2019).
- Haddad, Y.; Heger, Z.; Adam, V. Guidelines for Homology Modeling of Dopamine, Norepinephrine, and Serotonin Transporters. ACS Chem. Neurosci. 2016, 7, 1607–1613. [Google Scholar] [CrossRef]
- Boudker, O.; Verdon, G. Structural perspectives on secondary active transporters. Trends Pharmacol. Sci. 2013, 31, 418–426. [Google Scholar] [CrossRef] [Green Version]
- SWISS-MODEL Documentation. Available online: https://swissmodel.expasy.org/docs/help#model_results (accessed on 2 June 2019).
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Andersen, J.; Ringsted, K.B.; Bang-Andersen, B.; Strømgaard, K.; Kristensen, A.S. Binding site residues control inhibitor selectivity in the human norepinephrine transporter but not in the human dopamine transporter. Sci. Rep. 2015, 5, 15650. [Google Scholar] [CrossRef] [Green Version]
- Ravna, A.W.; Sylte, I.; Dahl, S.G. Structure and localisation of drug binding sites on neurotransmitter transporters. J. Mol. Model. 2009, 15, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Yamashita, A.; Gouaux, E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 2007, 448, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Nyola, A.; Karpowich, N.K.; Zhen, J.; Marden, J.; Reith, M.E.; Wang, D.N. Substrate and drug binding sites in LeuT. Curr. Opin. Struct. Biol. 2010, 20, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Zhen, J.; Karpowich, N.K.; Law, C.J.; Reith, M.E.A.; Wang, D.N. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 2009, 16, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.H.; Bahar, I. Molecular Mechanism of Dopamine Transport by Human Dopamine Transporter. Structure 2015, 23, 2171–2181. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.; Weissensteiner, R.; Steiner, I.; Sitte, H.H.; Ecker, G.F.; Freissmuth, M.; Sucic, S. The high-affinity binding site for tricyclic antidepressants resides in the outer vestibule of the serotonin transporter. Mol. Pharmacol. 2010, 78, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Zhu, L.; Lieberman, B.P.; Zha, Z.; Plössl, K.; Kung, H.F. Synthesis and evaluation of novel tropane derivatives as potential PET imaging agents for the dopamine transporter. Bioorg. Med. Chem. Lett. 2012, 22, 4303–4306. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.; Stuhr-Hansen, N.; Zachariassen, L.; Toubro, S.; Hansen, S.M.R.; Eildal, J.N.N.; Bond, A.D.; Bøgesø, K.P.; Bang-Andersen, B.; Kristensen, A.S.; et al. Molecular determinants for selective recognition of antidepressants in the human serotonin and norepinephrine transporters. Proc. Natl. Acad. Sci. USA 2011, 108, 12137–12142. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Goehring, A.; Wang, K.H.; Penmatsa, A.; Ressler, R.; Gouaux, E. Structural basis for action by diverse antidepressants on biogenic amine transporter. Nature 2013, 503, 141–145. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Knowledgebase. Available online: https://www.uniprot.org/uniprot/P23975 (accessed on 20 March 2018).
- Zhou, J.; He, R.; Johnson, K.M.; Ye, Y.; Kozikowski, A.P. Piperidine-based nocaine/modafinil hybrid ligands as highly potent monoamine transporter inhibitors: Efficient drug discovery by rational lead hybridization. J. Med. Chem. 2004, 47, 5821–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ChEMBL Database—EMBL-EBI. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 20 March 2018).
- Fensome, A.; Goldberg, J.; McComas, C.C.; Trybulski, E.J.; Woodworth, R.P.; Deecher, D.C.; Whiteside, G.T.; Zhang, P. Structure-activity relationships of norepinephrine reuptake inhibitors with benzothiadiazine dioxide or dihydrosulfostyril cores. Bioorg. Med. Chem. Lett. 2010, 20, 1555–1558. [Google Scholar] [CrossRef] [PubMed]
- Boot, J.; Cases, M.; Clark, B.P.; Findlay, J.; Gallagher, P.T.; Hayhurst, L.; Man, T.; Montalbetti, C.; Rathmell, R.E.; Rudyk, H.; et al. Discovery and structure-activity relationships of novel selective norepinephrine and dual serotonin/norepinephrine reuptake inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 699–703. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB. Available online: https://www.rcsb.org/structure/4xpg (accessed on 10 March 2018).
- RCSB PDB. Available online: https://www.rcsb.org/structure/4m48 (accessed on 10 March 2018).
- SWISS-MODEL. Available online: https://swissmodel.expasy.org (accessed on 10 March 2018).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | aLeuT (2qju) | hDAT [23] | dDAT (4m48) | hNET [23] | hNET Models 4xpg and 4m48 |
|---|---|---|---|---|---|
| Na+1 | leucine (substrate) | Asp79 | Asp46 via H2O | Asp75 | Asp75 |
| Ala22 | Ala77 | Ala44 | Ala73 | Ala73 | |
| Asn27 | Asn82 | Asn49 | Asn78 | Asn78 | |
| Thr254 | Ser321 | Ser320 | Ser318 | Ser318 | |
| Asn286 | Asn353 | Asn352 | Asn350 | Asn350 | |
| Na+2 | Gly20 | Gly75 | Gly42 | Gly71 | Gly71 |
| Val23 | Val78 | Val45 | Val74 | Val74 | |
| Ala351 | Leu418 | Leu417 | Leu415 | Leu415 | |
| Thr354 | Asp421 | Asp420 | Asp418 | Asp418 | |
| Ser355 | Ser422 | Ser421 | Ser419 | Ser419 | |
| Cl− | Lys121 | Tyr102 | Tyr69 | Tyr98 | Tyr98 |
| Ser150 | Ser321 | Ser320 | Ser318 | Ser318 | |
| Tyr151 | Asn353 | Asn352 | Asn350 | Asn350 | |
| Ser165 | Gln317 | Gln316 | Gln314 | Gln314 | |
| Phe167 | Ser357 | Ser356 | Ser354 | Ser354 |
| Compound | hNET Model 4xpg | hNET Model 4m48 |
|---|---|---|
| Desipramine | −7.602 | −9.225 |
| Nortriptyline | −7.137 | −9.432 |
| Reboxetine | −7.071 | −8.158 |
| Bupropion | −6.584 | −6.859 |
| Duloxetine | −7.859 | −7.295 |
| Mazindol | −8.845 | −8.624 |
| Modafinil | −7.793 | −8.177 |
| Nocaine | −7.392 | −7.269 |
| Venlafaxine | −7.603 | −7.674 |
| Compound X | −8.530 | −7.620 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Góral, I.; Łątka, K.; Bajda, M. Structure Modeling of the Norepinephrine Transporter. Biomolecules 2020, 10, 102. https://doi.org/10.3390/biom10010102
Góral I, Łątka K, Bajda M. Structure Modeling of the Norepinephrine Transporter. Biomolecules. 2020; 10(1):102. https://doi.org/10.3390/biom10010102
Chicago/Turabian StyleGóral, Izabella, Kamil Łątka, and Marek Bajda. 2020. "Structure Modeling of the Norepinephrine Transporter" Biomolecules 10, no. 1: 102. https://doi.org/10.3390/biom10010102
APA StyleGóral, I., Łątka, K., & Bajda, M. (2020). Structure Modeling of the Norepinephrine Transporter. Biomolecules, 10(1), 102. https://doi.org/10.3390/biom10010102