Generation of Retinaldehyde for Retinoic Acid Biosynthesis


Abstract
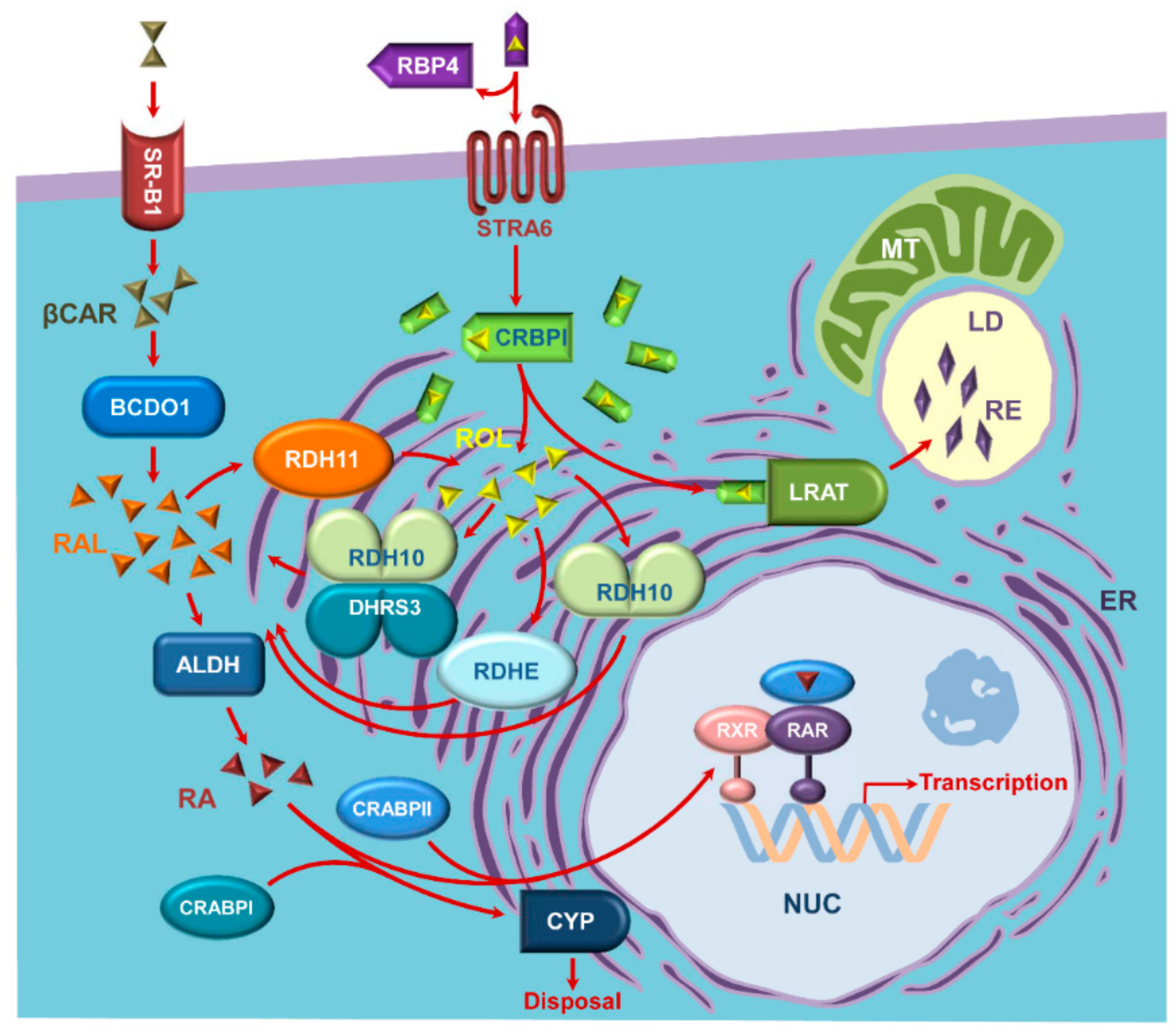
:1. Introduction
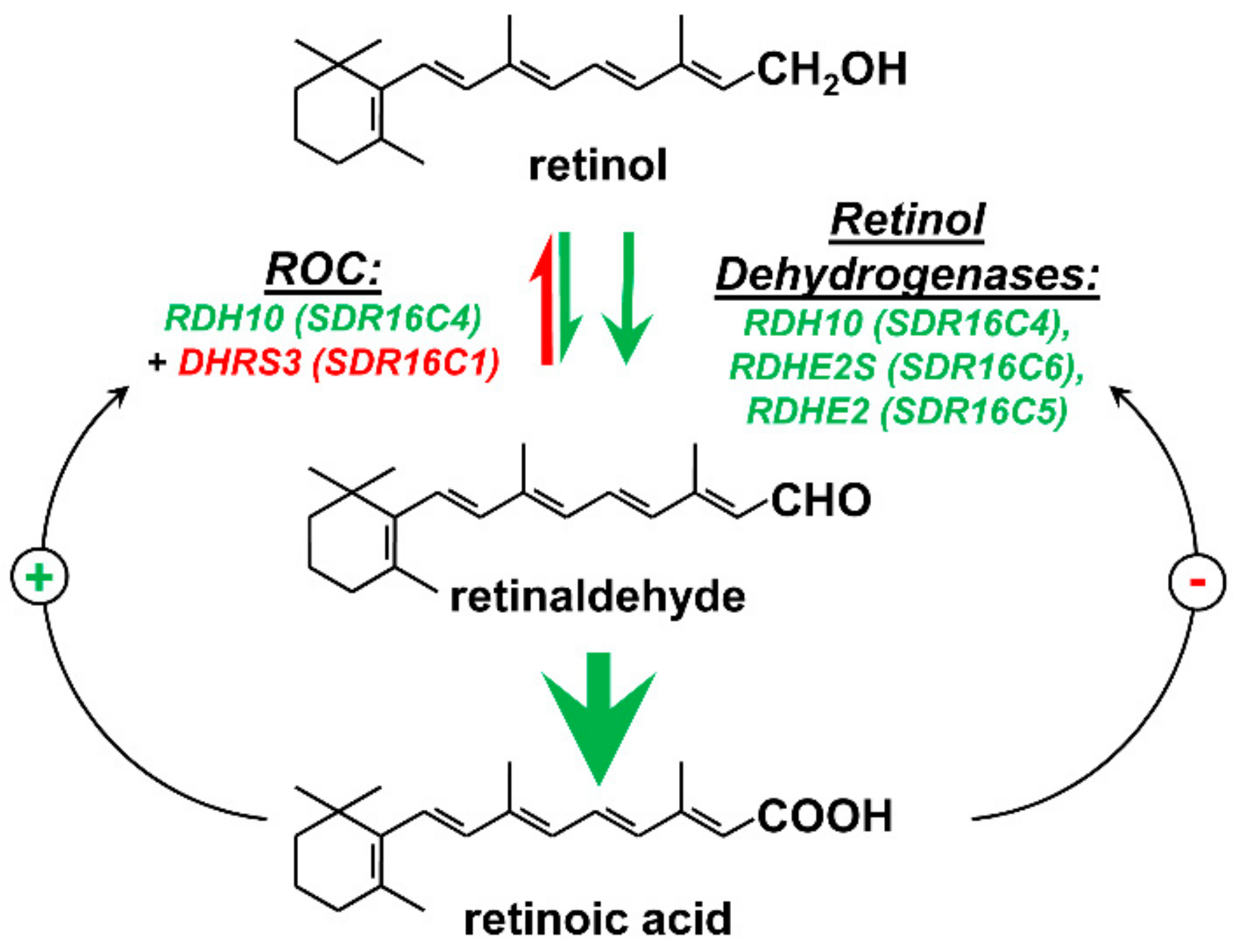
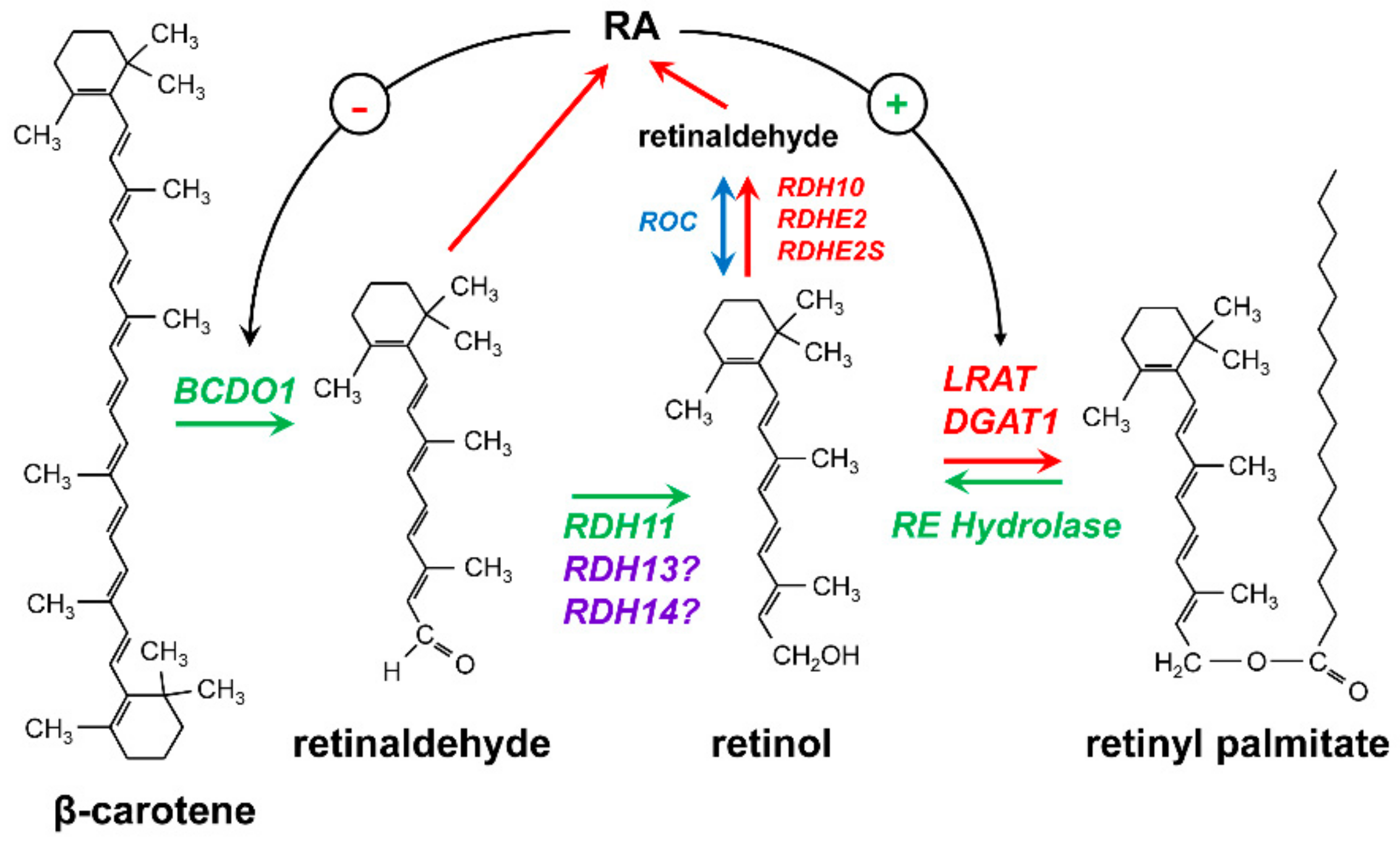
2. Retinoid-Active Members of the Short-Chain Dehydrogenase/Reductase Superfamily of Proteins
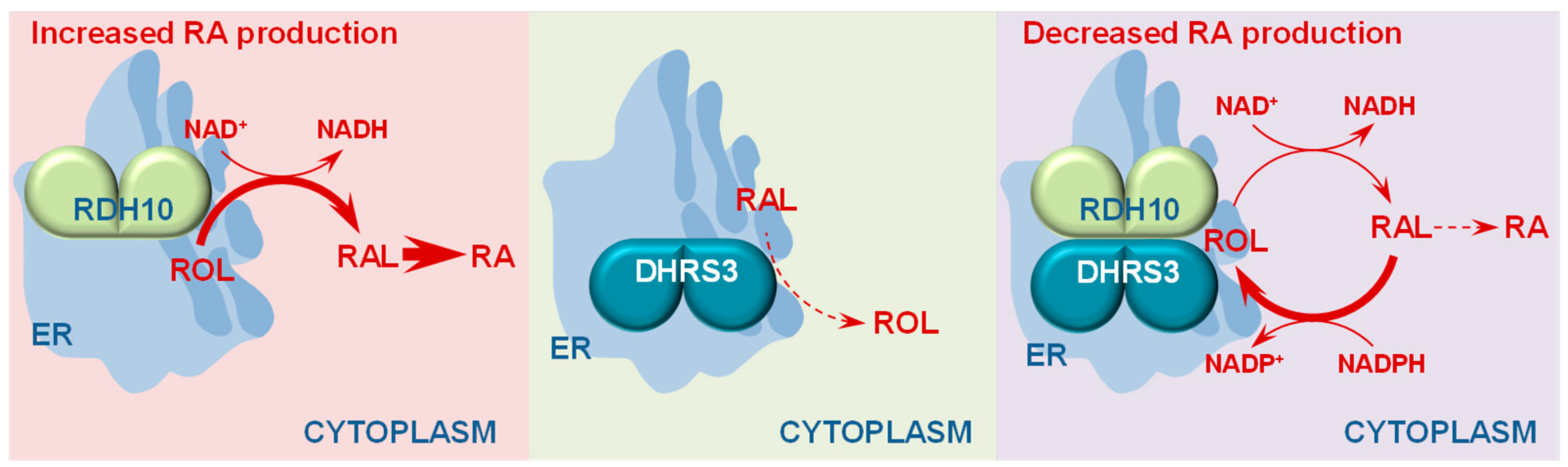
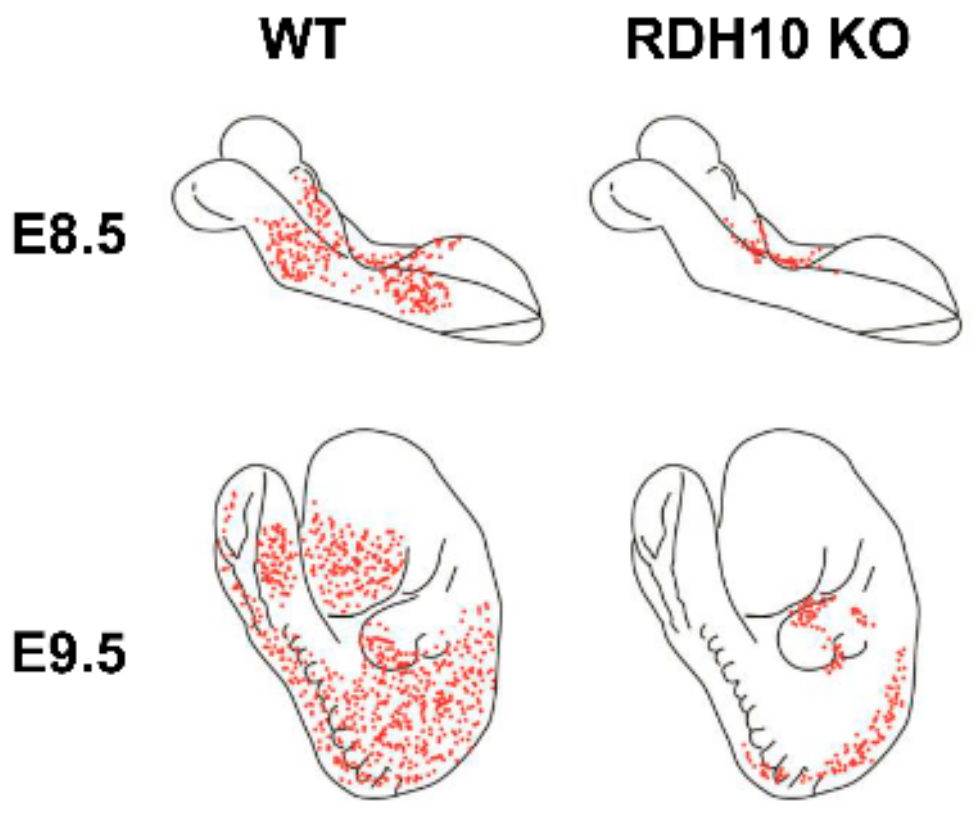
3. Retinol Dehydrogenase 10 (RDH10) and Regulation of the Flux from Retinol to Retinaldehyde
4. RDH10-Related Dehydrogenases
5. Retinol Dehydrogenase 11 and Maintenance of Retinol Homeostasis
6. RDH11-Related Enzymes
7. Future Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kedishvili, N.Y. Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, J.L. Retinol metabolism in UC-PKI cells. Characterization of retinoic acid synthesis by an established mammalian cell line. J. Biol. Chem. 1986, 261, 13592–13597. [Google Scholar] [PubMed]
- McCaffrery, P.; Posch, K.C.; Napoli, J.L.; Gudas, L.; Dräger, U.C. Changing patterns of the retinoic acid system in the developing retina. Dev. Biol. 1993, 158, 390–399. [Google Scholar] [CrossRef] [PubMed]
- McCaffery, P.; Dräger, U.C. Retinoic acid synthesizing enzymes in the embryonic and adult vertebrate. Adv. Exp. Med. Biol. 1995, 372, 173–183. [Google Scholar] [PubMed]
- McCaffery, P.; Dräger, U.C. A sensitive bioassay for enzymes that synthesize retinoic acid. Brain Res. Brain Res. Protoc. 1997, 1, 232–236. [Google Scholar] [CrossRef]
- Wang, X.; Penzes, P.; Napoli, J.L. Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. J. Biol. Chem. 1996, 271, 16288–16293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; McCaffery, P.; Ivins, K.J.; Neve, R.L.; Hogan, P.; Chin, W.W.; Dräger, U.C. Molecular identification of a major retinoic-acid-synthesizing enzyme, a retinaldehyde-specific dehydrogenase. Eur. J. Biochem. 1996, 240, 15–22. [Google Scholar] [CrossRef]
- Gruin, F.; Hirose, Y.; Kawauchi, S.; Ogura, T.; Umesono, K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominantly expressed in sensory neuroepithelia during development. J. Biol. Chem. 2000, 275, 41210–41218. [Google Scholar] [CrossRef] [Green Version]
- Rossant, J.; Zirngibl, R.; Cado, D.; Shago, M.; Giguère, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991, 5, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Niederreither, K.; Fraulob, V.; Garnier, J.M.; Chambon, P.; Dollé, P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech. Dev. 2002, 110, 165–171. [Google Scholar] [CrossRef]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhinn, M.; Schuhbaur, B.; Niederreither, K.; Dollé, P. Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. USA 2011, 108, 16687–16692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farjo, K.M.; Moiseyev, G.; Nikolaeva, O.; Sandell, L.L.; Trainor, P.A.; Ma, J.X. RDH10 is the primary enzyme responsible for the first step of embryonic Vitamin A metabolism and retinoic acid synthesis. Dev. Biol. 2011, 357, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammas, L.; Romand, R.; Fraulob, V.; Mura, C.; Dollé, P. Expression of the murine retinol dehydrogenase 10 (Rdh10) gene correlates with many sites of retinoid signalling during embryogenesis and organ differentiation. Dev. Dyn. 2007, 236, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.; Kallberg, Y.; Bray, J.E.; Bruford, E.; Dellaporta, S.L.; Favia, A.D.; Duarte, R.G.; Jörnvall, H.; Kavanagh, K.L.; Kedishvili, N.; et al. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem. Biol. Interact. 2009, 178, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedishvili, N.Y. Retinoic Acid Synthesis and Degradation. Subcell. Biochem. 2016, 81, 127–161. [Google Scholar]
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 1969, 115, 609–619. [Google Scholar] [CrossRef]
- Haeseleer, F.; Huang, J.; Lebioda, L.; Saari, J.C.; Palczewski, K. Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. J. Biol. Chem. 1998, 273, 21790–21799. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.X.; Chen, Y.; Chen, Y.; Fan, J.; Rohrer, B.; Crouch, R.K.; Ma, J.X. Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3365–3372. [Google Scholar]
- Wu, B.X.; Moiseyev, G.; Chen, Y.; Rohrer, B.; Crouch, R.K.; Ma, J.X. Identification of RDH10, an All-trans Retinol Dehydrogenase, in Retinal Muller Cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3857–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaeva, O.V.; Johnson, M.P.; Kedishvili, N.Y. Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J. Biol. Chem. 2008, 283, 20299–20308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.A.; Belyaeva, O.V.; Wu, L.; Kedishvili, N.Y. Retinol dehydrogenase 10 but not retinol/sterol dehydrogenase(s) regulates the expression of retinoic acid-responsive genes in human transgenic skin raft culture. J. Biol. Chem. 2011, 286, 13550–13560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego, O.; Belyaeva, O.V.; Porté, S.; Ruiz, F.X.; Stetsenko, A.V.; Shabrova, E.V.; Kostereva, N.V.; Farrés, J.; Parés, X.; Kedishvili, N.Y. Comparative functional analysis of human medium-chain dehydrogenases, short-chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem. J. 2006, 399, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Romand, R.; Kondo, T.; Cammas, L.; Hashino, E.; Dollé, P. Dynamic expression of the retinoic acid-synthesizing enzyme retinol dehydrogenase 10 (rdh10) in the developing mouse brain and sensory organs. J. Comp. Neurol. 2008, 508, 879–892. [Google Scholar] [CrossRef]
- Sandell, L.L.; Lynn, M.L.; Inman, K.E.; McDowell, W.; Trainor, P.A. RDH10 oxidation of vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS ONE 2012, 7, e30698. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Chatzi, C.; Sandell, L.L.; Trainor, P.A.; Duester, G. Rdh10 mutants deficient in limb field retinoic acid signaling exhibit normal limb patterning but display interdigital webbing. Dev. Dyn. 2011, 240, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Ashique, A.M.; May, S.R.; Kane, M.A.; Folias, A.; Phamluong, E.K.; Choe, Y.; Napoli, J.L.; Peterson, A.S. Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis 2012, 50, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.-H.; Yang, Q.-E.; Davis, J.C.; Griswold, M.D. Retinol dehydrogenase 10 is indispensible for spermatogenesis in juvenile males. Proc. Natl. Acad. Sci. USA 2013, 110, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Hernandez, R.E.; Waxman, J.S.; Yelon, D.; Moens, C.B. Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Dev. Biol. 2010, 338, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Billings, S.E.; Pierzchalski, K.; Butler Tjaden, N.E.; Pang, X.Y.; Trainor, P.A.; Kane, M.A.; Moise, A.R. The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J. 2013, 27, 4877–4889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.K.; Belyaeva, O.V.; Wu, L.; Kedishvili, N.Y. The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 2014, 289, 14868–14880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaeva, O.V.; Stetsenko, A.V.; Nelson, P.; Kedishvili, N.Y. Properties of short-chain dehydrogenase/reductase RalR1: Characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry 2003, 42, 14838–14845. [Google Scholar] [CrossRef] [PubMed]
- Belyaeva, O.V.; Korkina, O.V.; Stetsenko, A.V.; Kim, T.; Nelson, P.S.; Kedishvili, N.Y. Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): Catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry 2005, 44, 7035–7047. [Google Scholar] [PubMed] [Green Version]
- Belyaeva, O.V.; Adams, M.K.; Wu, L.; Kedishvili, N.Y. The antagonistically bifunctional retinoid oxidoreductase complex is required for maintenance of all-trans-retinoic acid homeostasis. J. Biol. Chem. 2017, 292, 5884–5897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerignoli, F.; Guo, X.; Cardinali, B.; Rinaldi, C.; Casaletto, J.; Frati, L.; Screpanti, I.; Gudas, L.J.; Gulino, A.; Thiele, C.J.; et al. retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines. Cancer Res. 2002, 62, 1196–1204. [Google Scholar] [PubMed]
- Zolfaghari, R.; Chen, Q.; Ross, A.C. DHRS3, a retinal reductase, is differentially regulated by retinoic acid and lipopolysaccharide-induced inflammation in THP-1 cells and rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G578–G588. [Google Scholar] [CrossRef] [Green Version]
- Hart, Y.; Alon, U. The utility of paradoxical components in biological circuits. Mol. Cell 2013, 49, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Kedishvili, N.Y.; Belyaeva, O.V. Retinyl esters are elevated in progeny of retinol dehydrogenase 11 deficient dams. Chem. Biol. Interact. 2019, 302, 117–122. [Google Scholar] [CrossRef]
- Wang, S.; Huang, W.; Castillo, H.A.; Kane, M.A.; Xavier-Neto, J.; Trainor, P.A.; Moise, A.R. Alterations in retinoic acid signaling affect the development of the mouse coronary vasculature. Dev. Dyn. 2018, 247, 976–991. [Google Scholar] [CrossRef] [Green Version]
- Shannon, S.R.; Moise, A.R.; Trainor, P.A. New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, R.K.; Shi, W.; Chan, S.O.; Chen, Y.; Xu, G.; Lau, C.B.; Fung, K.P.; Chan, W.Y.; Zhao, H. Dhrs3 protein attenuates retinoic acid signaling and is required for early embryonic patterning. J. Biol. Chem. 2013, 288, 31477–31487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.J.; Nie, Q.; Lander, A.D.; Schilling, T.F. Complex regulation of cyp26a1 creates a robust retinoic acid gradient in the zebrafish embryo. PLoS Biol. 2007, 5, e304. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.Q.; Radtke, K.; Linville, A.; Lander, A.D.; Nie, Q.; Schilling, T.F. Cellular retinoic acid-binding proteins are essential for hindbrain patterning and signal robustness in zebrafish. Development 2012, 139, 2150–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, T.F.; Nie, Q.; Lander, A.D. Dynamics and precision in retinoic acid morphogen gradients. Curr. Opin. Genet. Dev. 2012, 22, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Brade, T.; Sandell, L.L.; Lewandoski, M.; Trainor, P.A.; Colas, A.; Mercola, M.; Duester, G. Retinoic Acid Activity in Undifferentiated Neural Progenitors Is Sufficient to Fulfill Its Role in Restricting Fgf8 Expression for Somitogenesis. PLoS ONE 2015, 10, e0137894. [Google Scholar] [CrossRef]
- Belyaeva, O.V.; Lee, S.A.; Adams, M.K.; Chang, C.; Kedishvili, N.Y. Short chain dehydrogenase/reductase rdhe2 is a novel retinol dehydrogenase essential for frog embryonic development. J. Biol. Chem. 2012, 287, 9061–9071. [Google Scholar] [CrossRef] [Green Version]
- Strate, I.; Min, T.H.; Iliev, D.; Pera, E.M. Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development 2009, 136, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Koide, T.; Downes, M.; Chandraratna, R.A.; Blumberg, B.; Umesono, K. Active repression of RAR signaling is required for head formation. Genes Dev. 2001, 15, 2111–2121. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.; Luo, T.; Dräger, U.C. Retinoic acid synthesis in the postnatal mouse brain marks distinct developmental stages and functional systems. Cereb. Cortex 2002, 12, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Belyaeva, O.V.; Adams, M.K.; Klyuyeva, A.V.; Lee, S.A.; Goggans, K.R.; Kesterson, R.A.; Popov, K.M.; Kedishvili, N.Y. Mice lacking the epidermal retinol dehydrogenases SDR16C5 and SDR16C6 display accelerated hair growth and enlarged meibomian glands. J. Biol. Chem. 2019, 294, 17060–17074. [Google Scholar] [CrossRef] [PubMed]
- Belyaeva, O.V.; Chang, C.; Berlett, M.C.; Kedishvili, N.Y. Evolutionary origins of retinoid active short-chain dehydrogenases/reductases of SDR16C family. Chem. Biol. Interact. 2015, 234, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.A.; Belyaeva, O.V.; Kedishvili, N.Y. Biochemical characterization of human epidermal retinol dehydrogenase 2. Chem. Biol. Interact. 2009, 178, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.K.; Lee, S.A.; Belyaeva, O.V.; Wu, L.; Kedishvili, N.Y. Characterization of human short chain dehydrogenase/reductase SDR16C family members related to retinol dehydrogenase 10. Chem. Biol. Interact. 2017, 276, 88–94. [Google Scholar] [CrossRef]
- Pryce, J.E.; Hayes, B.J.; Bolormaa, S.; Goddard, M.E. Polymorphic regions affecting human height also control stature in cattle. Genetics 2011, 187, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, S.; Watanabe, T.; Mizoshita, K.; Tatsuda, K.; Fujita, T.; Watanabe, N.; Sugimoto, Y.; Takasuga, A. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Walters, G.B.; Thorleifsson, G.; Stefansson, H.; Halldorsson, B.V.; Zusmanovich, P.; Sulem, P.; Thorlacius, S.; Gylfason, A.; Steinberg, S.; et al. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008, 40, 609–615. [Google Scholar] [CrossRef]
- Weedon, M.N.; Lango, H.; Lindgren, C.M.; Wallace, C.; Evans, D.M.; Mangino, M.; Freathy, R.M.; Perry, J.R.B.; Stevens, S.; Hall, A.S.; et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008, 40, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Lettre, G.; Jackson, A.U.; Gieger, C.; Schumacher, F.R.; Berndt, S.I.; Sanna, S.; Eyheramendy, S.; Voight, B.F.; Butler, J.L.; Guiducci, C.; et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008, 40, 584–591. [Google Scholar] [CrossRef]
- Karim, L.; Takeda, H.; Lin, L.; Druet, T.; Arias, J.A.C.; Baurain, D.; Cambisano, N.; Davis, S.R.; Farnir, F.; Grisart, B.; et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 2011, 43, 405–413. [Google Scholar] [CrossRef]
- Littlejohn, M.; Grala, T.; Sanders, K.; Walker, C.; Waghorn, G.; Macdonald, K.; Coppieters, W.; Georges, M.; Spelman, R.; Hillerton, E.; et al. Genetic variation in PLAG1 associates with early life body weight and peripubertal weight and growth in Bos Taurus. Anim. Genet. 2012, 43, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Maltecca, C.; Gray, K.A.; Cassady, J.P. Feed intake, average daily gain, feed efficiency, and real-time ultrasound traits in Duroc pigs: II. Genomewide association. J. Anim. Sci. 2014, 92, 2846–2860. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhu, J.; Sun, Y.; Liu, R.; Liu, N.; Li, D.; Wen, J.; Chen, J. Identification of genes related to beak deformity of chickens using digital gene expression profiling. PLoS ONE 2014, 9, e107050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Yang, H.; Yang, B.; Chen, C.; Huang, L. Identification of quantitative trait transcripts for growth traits in the large scales of liver and muscle samples. Physiol. Genom. 2015, 47, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; White, J.T.; Ferguson, C.; Wang, S.; Vessella, R.; Bumgarner, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate short-chain dehydrogenase reductase 1 (PSDR1): A new member of the short-chain steroid dehydrogenase/reductase family highly expressed in normal and neoplastic prostate epithelium. Cancer Res. 2001, 61, 1611–1618. [Google Scholar]
- Kedishvili, N.Y.; Chumakova, O.V.; Chetyrkin, S.V.; Belyaeva, O.V.; Lapshina, E.A.; Lin, D.W.; Matsumura, M.; Nelson, P.S. Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1). J. Biol. Chem. 2002, 277, 28909–28915. [Google Scholar] [CrossRef] [Green Version]
- Kasus-Jacobi, A.; Ou, J.; Bashmakov, Y.K.; Shelton, J.M.; Richardson, J.A.; Goldstein, J.L.; Brown, M.S. Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2003, 278, 32380–32389. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.S.; Maeda, A.; Maeda, T.; Heinlein, C.; Kedishvili, N.; Palczewski, K.; Nelson, P.S. Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: A role of RDH11 in visual processes in vivo. J. Biol. Chem. 2005, 280, 8694–8704. [Google Scholar] [CrossRef] [Green Version]
- Kasus-Jacobi, A.; Ou, J.; Birch, D.G.; Locke, K.G.; Shelton, J.M.; Richardson, J.A.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Edwards, A.O. Functional characterization of mouse RDH11 as a retinol dehydrogenase involved in dark adaptation in vivo. J. Biol. Chem. 2005, 280, 20413–20420. [Google Scholar] [CrossRef] [Green Version]
- Marchette, L.D.; Thompson, D.A.; Kravtsova, M.; Ngansop, T.N.; Mandal, M.N.; Kasus-Jacobi, A. Retinol dehydrogenase 12 detoxifies 4-hydroxynonenal in photoreceptor cells. Free Radic. Biol. Med. 2010, 48, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Kanan, Y.; Wicker, L.D.; Al-Ubaidi, M.R.; Mandal, N.A.; Kasus-Jacobi, A. Retinol dehydrogenases RDH11 and RDH12 in the mouse retina: Expression levels during development and regulation by oxidative stress. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Shen, Y. Mouse ENCODE Consortium. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunskill, E.W.; Potter, A.S.; Distasio, A.; Dexheimer, P.; Plassard, A.; Aronow, B.J.; Potter, S.S. A gene expression atlas of early craniofacial development. Dev. Biol. 2014, 391, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaeva, O.V.; Wu, L.; Shmarakov, I.; Nelson, P.S.; Kedishvili, N.Y. Retinol dehydrogenase 11 is essential for the maintenance of retinol homeostasis in liver and testis in mice. J. Biol. Chem. 2018, 293, 6996–7007. [Google Scholar] [CrossRef] [Green Version]
- Von Lintig, J. Provitamin a metabolism and functions in mammalian biology. Am. J. Clin. Nutr. 2012, 96, 1234S–1244S. [Google Scholar] [CrossRef] [Green Version]
- Harrison, E.H. Mechanisms of digestion and absorption of dietary vitamin A. Annu. Rev. Nutr. 2005, 25, 87–103. [Google Scholar] [CrossRef]
- Blaner, W.S.; Li, Y.; Brun, P.J.; Yuen, J.J.; Lee, S.A.; Clugston, R.D. Vitamin A Absorption, Storage and Mobilization. Subcell. Biochem. 2016, 81, 95–125. [Google Scholar]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Mungall, C.J. FANTOM consortium. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Belyaeva, O.V.; Kedishvili, N.Y. Human pancreas protein 2 (PAN2) has a retinal reductase activity and is ubiquitously expressed in human tissues. FEBS Lett. 2002, 531, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Belyaeva, O.V.; Korkina, O.V.; Stetsenko, A.V.; Kedishvili, N.Y. Human retinol dehydrogenase 13 (RDH13) is a mitochondrial short-chain dehydrogenase/reductase with a retinaldehyde reductase activity. FEBS J. 2008, 275, 138–147. [Google Scholar] [CrossRef]
- Keller, B.; Adamski, J. RDH12, a retinol dehydrogenase causing Leber’s congenital amaurosis, is also involved in steroid metabolism. J. Steroid Biochem. Mol. Biol. 2007, 104, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Isken, A.; Hoff, S.; Babino, D.; von Lintig, J. BCDO2 acts as a carotenoid scavenger and gatekeeper for the mitochondrial apoptotic pathway. Development 2012, 139, 2966–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cui, X.; Gu, Q.; Chen, Y.; Zhou, J.; Kuang, Y.; Wang, Z.; Xu, X. Retinol dehydrogenase 13 protects the mouse retina from acute light damage. Mol. Vis. 2012, 18, 1021–1030. [Google Scholar] [PubMed]
- Cui, X.; Dang, S.; Wang, Y.; Chen, Y.; Zhou, J.; Shen, C.; Kuang, Y.; Fei, J.; Lu, L.; Wang, Z. Retinol dehydrogenase 13 deficiency diminishes carbon tetrachloride-induced liver fibrosis in mice. Toxicol. Lett. 2017, 265, 17–22. [Google Scholar] [CrossRef]
- Cui, X.; Ma, B.; Wang, Y.; Chen, Y.; Shen, C.; Kuang, Y.; Fei, J.; Lu, L.; Wang, Z. Rdh13 deficiency weakens carbon tetrachloride-induced liver injury by regulating Spot14 and Cyp2e1 expression levels. Front Med. 2019, 13, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Jang, G.F.; Imanishi, Y.; Driessen, C.A.G.G.; Matsumura, M.; Nelson, P.S.; Palczewski, K. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 2002, 277, 45537–45546. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Miyazono, S.; Tachibanaki, S.; Kawamura, S. RDH13L, an enzyme responsible for the aldehyde-alcohol redox coupling reaction (AL-OL coupling reaction) to supply 11-cis retinal in the carp cone retinoid cycle. J. Biol. Chem. 2015, 290, 2983–2992. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.C.; Jacobs, H.; Marwarha, G.; Gely-Pernot, A.; O’Byrne, S.M.; DeSantis, D.; Klopfenstein, M.; Feret, B.; Dennefeld, C.; Blaner, W.S.; et al. The STRA6 Receptor Is Essential for Retinol-Binding Protein-Induced Insulin Resistance but Not for Maintaining Vitamin A Homeostasis in Tissues Other Than the Eye. J. Biol. Chem. 2013, 288, 24528–24539. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Napoli, J.L. The Retinol Dehydrogenase Rdh10 Localizes to Lipid Droplets during Acyl Ester Biosynthesis. J. Biol. Chem. 2013, 288, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Deisenroth, C.; Itahana, Y.; Tollini, L.; Jin, A.; Zhang, Y. p53-inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. J. Biol. Chem. 2011, 286, 28343–28356. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sandell, L.L.; Trainor, P.A.; Koentgen, F.; Duester, G. Alcohol and aldehyde dehydrogenases: Retinoid metabolic effects in mouse knockout models. Biochim. Biophys. Acta 2011, 1821, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, M.A.; Sandell, L.L. Enzymatic Metabolism of Vitamin A in Developing Vertebrate Embryos. Nutrients 2016, 8, 812. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SDR | Substrate/Cofactor | Apparent Km μM | Apparent Vmax nmol⋅min−1⋅mg−1 | Vmax/Km |
|---|---|---|---|---|
| Xenopus rdhe251 | all-trans-retinol | 0.6 ± 0.1 | 19.5 ± 0.6 | 32.5 |
| NAD+ | 108 ± 27 | 21 ± 1 | ||
| all-trans-retinaldehyde | 0.6 ± 0.1 | 3.6 ± 0.1 | 6 | |
| NADH | 8.4 ± 1.7 | 4.1 ± 0.2 | ||
| 11-cis-retinol | 3.3 ± 0.4 | 3.9 ± 0.2 | 1.8 | |
| Murine RDHE251 | all-trans-retinol | UD | UD | |
| all-trans-retinaldehyde | N.D. | N.D. | ||
| Murine RDHE2S51 | all-trans-retinol | 0.87 ± 0.21 | 8.7 ± 0.6 | 10 |
| NAD+ | 460 ± 30 | 5.8 ± 0.2 | ||
| all-trans-retinaldehyde | 0.6 ± 0.1 | 3.6 ± 0.1 | 6 | |
| NADH | 11 ± 3 | 2.6 ± 0.1 | ||
| 11-cis-retinol | 0.86 ± 0.14 | 1.34 ± 0.07 | 1.6 | |
| 9-cis-retinol | UD | UD | ||
| Human RDHE253 | all-trans-retinol | N.D. | ~0.06 | |
| NAD+ | Preferred | |||
| Human RDH1022 | all-trans-retinol | 0.035 ± 0.010 | 1.30 ± 0.05 | 37 |
| NAD+ | 100 ± 10 | 1.30 ± 0.04 | ||
| 11-cis-retinol | 0.06 ± 0.01 | 1.42 ± 0.06 | 24 | |
| 9-cis-retinol | 0.04 ± 0.01 | 0.77 ± 0.05 | 19 |
| SDR | Substrate/Cofactor | Apparent Km μM | Apparent Vmax nmol·min−1·mg−1 | Vmax/Km | kcat min−1 | kcat/Km min−1·μM−1 |
|---|---|---|---|---|---|---|
| RDH11 Sf9 MS33 | all-trans-retinol | 0.70 ± 0.04 | 13.2 ± 0.3 | 19 | ||
| NADP+ | 0.4 ± 0.2 | 8.7 ± 0.8 | ||||
| all-trans-retinaldehyde | 0.20 ± 0.01 | 41 ± 1 | 205 | |||
| NADPH | 0.48 ± 0.02 | 44.0 ± 0.6 | ||||
| RDH11 Purified33 | all-trans-retinol | 0.6 ± 0.1 | 300 ± 20 | 500 | 11 | 18 |
| NADP+ | 1.0 ± 0.1 | 250 ± 4 | ||||
| all-trans-retinaldehyde | 0.12 ± 0.1 | 506 ± 13 | 4217 | 18 | 150 | |
| NADPH | 0.47 ± 0.04 | 530 ± 10 | ||||
| RDH12, Sf9 MS34 | NADP+ (with atROL) | 1.2 ± 0.2 | 79.0 ± 2.5 | |||
| NADPH (with atRAL) | 1.2 ± 0.3 | 98.0 ± 2.0 | ||||
| RDH12 Purified34 | all-trans-retinol | 0.40 ± 0.10 | 748 | 1870 | 27 ± 2 | 68 |
| 11-cis-retinol | 0.16 ± 0.03 | 7 ± 0.7 | 44 | |||
| 9-cis-retinol | 0.16 ± 0.03 | 7 ± 0.3 | 44 | |||
| NADP+ (with atROL) | 3.2 ± 0.2 | 770 ± 30 | ||||
| all-trans-retinaldehyde | 0.04 ± 0.01 | 997 | 24,930 | 36 ± 1.7 | 900 | |
| 11-cis-retinaldehyde | 0.1 ± 0.05 | 45 ± 4 | 450 | |||
| 9-cis-retinaldehyde | 0.14 ± 0.03 | 14 ± 1 | 100 | |||
| NADPH (with atRAL) | 0.74 ± 0.06 | 1020 ± 20 | ||||
| RDH13 Purified80 | all-trans-retinol | ~ 3 | ~ 5 | |||
| NADP+ | ~6000 | ~25 | ||||
| all-trans-retinaldehyde | 3.2 ± 0.7 | 230 ± 24 | 72 | 8.2 | 2.6 | |
| NADPH | 1.5 ± 0.1 | 230 ± 24 | ||||
| RDH14 Sf9 MS79 | all-trans-retinol | 0.4 ± 0.07 | 31 ± 2 | 78 | ||
| NADP+ | 0.65 ± 0.11 | |||||
| all-trans-retinaldehyde | 0.08 ± 0.2 | 27 ± 1 | 338 | |||
| NADPH | 0.32 ± 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belyaeva, O.V.; Adams, M.K.; Popov, K.M.; Kedishvili, N.Y. Generation of Retinaldehyde for Retinoic Acid Biosynthesis. Biomolecules 2020, 10, 5. https://doi.org/10.3390/biom10010005
Belyaeva OV, Adams MK, Popov KM, Kedishvili NY. Generation of Retinaldehyde for Retinoic Acid Biosynthesis. Biomolecules. 2020; 10(1):5. https://doi.org/10.3390/biom10010005
Chicago/Turabian StyleBelyaeva, Olga V., Mark K. Adams, Kirill M. Popov, and Natalia Y. Kedishvili. 2020. "Generation of Retinaldehyde for Retinoic Acid Biosynthesis" Biomolecules 10, no. 1: 5. https://doi.org/10.3390/biom10010005
APA StyleBelyaeva, O. V., Adams, M. K., Popov, K. M., & Kedishvili, N. Y. (2020). Generation of Retinaldehyde for Retinoic Acid Biosynthesis. Biomolecules, 10(1), 5. https://doi.org/10.3390/biom10010005