
Sequence Features of Mitochondrial Transporter Protein Families


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Trafficking of Membrane Transporters into Mitochondria

2. SLC25—Mitochondrial Carrier Family (MCF)
3. SLC54—Mitochondrial Pyruvate Carriers (MPC)
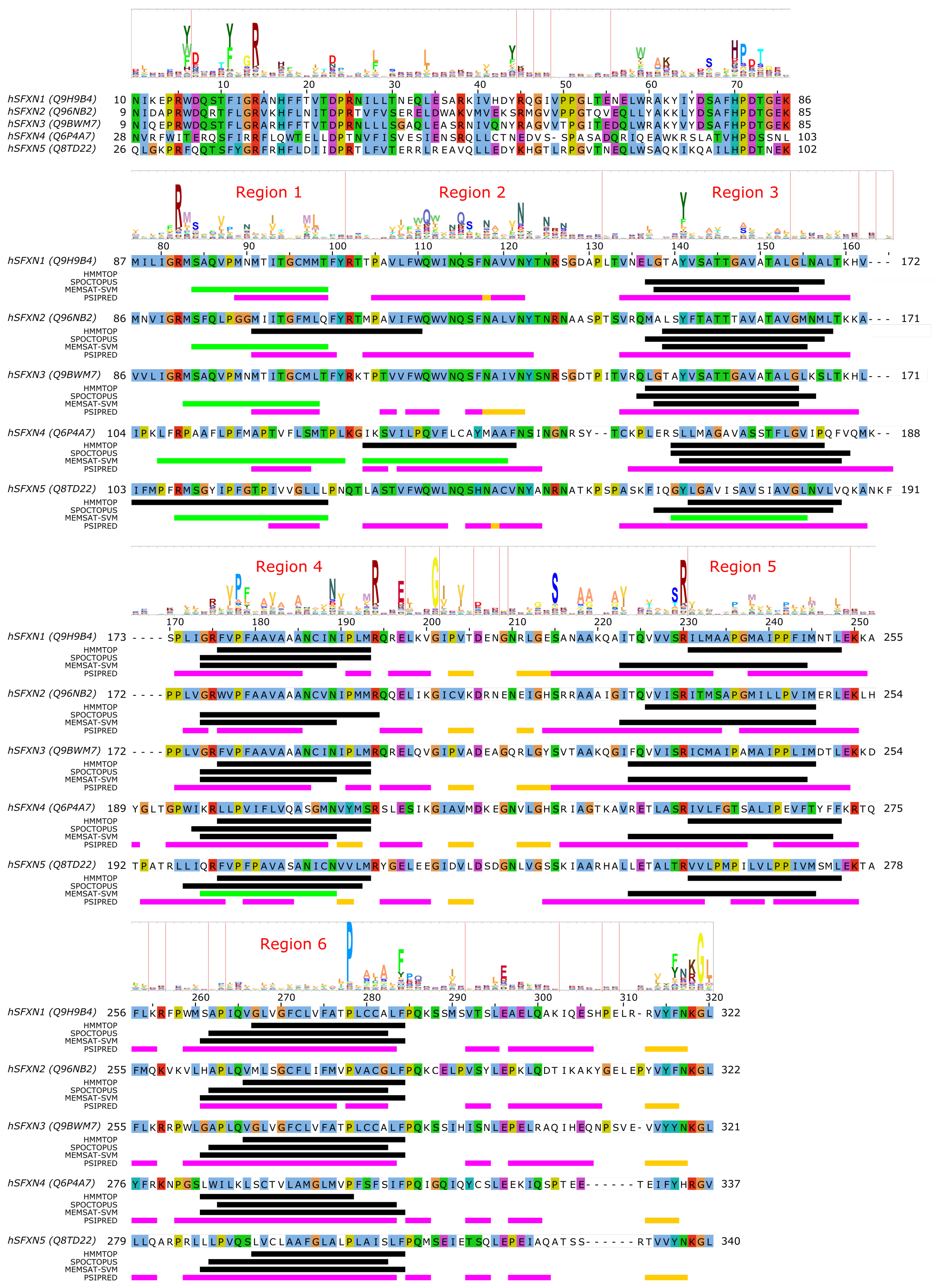
4. SLC56—Sideroflexins
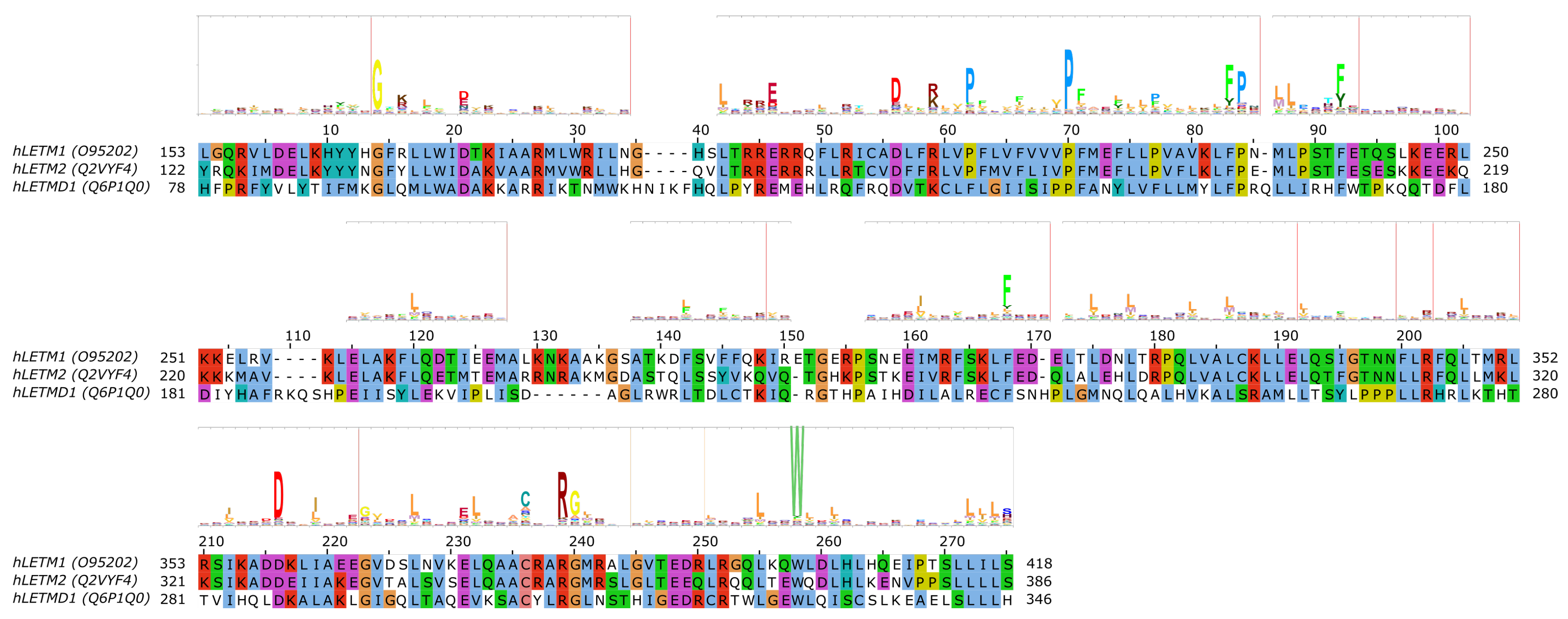
5. The SLC55/LETM Mitochondrial Cation/Proton Exchanger Family
6. ATP-Binding Cassette (ABC) Transporters in Mitochondria
7. Mitochondrial Calcium Transport via SLC8 Family
8. Additional Families with Members Proposed to Be Localized in the IMM
8.1. The SLC9 Na+/H+ Exchanger Family
8.2. The SLC1 Glutamate/Neutral Amino Acid Transporter Family
9. Conclusions and Open Questions
Funding
Conflicts of Interest
References
- Koonin, E.V. The Origin and Early Evolution of Eukaryotes in the Light of Phylogenomics. Genome Biol. 2010, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicheritz-Pontén, T.; Kurland, C.G.; Andersson, S.G. A Phylogenetic Analysis of the Cytochrome b and Cytochrome c Oxidase I Genes Supports an Origin of Mitochondria from within the Rickettsiaceae. Biochim. Biophys. Acta 1998, 1365, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Kurland, C.G.; Andersson, S.G. Origin and Evolution of the Mitochondrial Proteome. Microbiol. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar] [CrossRef] [Green Version]
- Karlberg, O.; Canbäck, B.; Kurland, C.G.; Andersson, S.G. The Dual Origin of the Yeast Mitochondrial Proteome. Yeast 2000, 17, 170–187. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.W.; Lang, B.F.; Cedergren, R.; Golding, G.B.; Lemieux, C.; Sankoff, D.; Turmel, M.; Brossard, N.; Delage, E.; Littlejohn, T.G.; et al. Genome Structure and Gene Content in Protist Mitochondrial DNAs. Nucleic Acids Res. 1998, 26, 865–878. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Vothknecht, U.C.; Szabo, I. Mitochondrial Ion Channels and Transporters in Plants: Prediction and Facts. Mitochondrion 2020, 53, 224–233. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. Bioessays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1800013. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokranjac, D.; Neupert, W. Cell Biology: Architecture of a Protein Entry Gate. Nature 2015, 528, 201–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular Architecture of the Active Mitochondrial Protein Gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- von Heijne, G.; Steppuhn, J.; Herrmann, R.G. Domain Structure of Mitochondrial and Chloroplast Targeting Peptides. Eur. J. Biochem. 1989, 180, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.G.; Gould, S.B. The Role of Charge in Protein Targeting Evolution. Trends Cell Biol. 2016, 26, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Shodai, T.; Muto, T.; Mihara, K.; Torii, H.; Nishikawa, S.; Endo, T.; Kohda, D. Structural Basis of Presequence Recognition by the Mitochondrial Protein Import Receptor Tom20. Cell 2000, 100, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Mokranjac, D.; Neupert, W. The Many Faces of the Mitochondrial TIM23 Complex. Biochim. Biophys. Acta 2010, 1797, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Ieva, R.; Schrempp, S.G.; Opaliński, L.; Wollweber, F.; Höß, P.; Heißwolf, A.K.; Gebert, M.; Zhang, Y.; Guiard, B.; Rospert, S.; et al. Mgr2 Functions as Lateral Gatekeeper for Preprotein Sorting in the Mitochondrial Inner Membrane. Mol. Cell 2014, 56, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Steffen, J.; Koehler, C.M. The Great Escape: Mgr2 of the Mitochondrial TIM23 Translocon Is a Gatekeeper Tasked with Releasing Membrane Proteins. Mol. Cell 2014, 56, 613–614. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, M.; Rehling, P.; Guiard, B.; Herrmann, J.M.; Pfanner, N.; van der Laan, M. Cooperation of Stop-Transfer and Conservative Sorting Mechanisms in Mitochondrial Protein Transport. Curr. Biol. 2010, 20, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- van der Giezen, M.; Slotboom, D.J.; Horner, D.S.; Dyal, P.L.; Harding, M.; Xue, G.P.; Embley, T.M.; Kunji, E.R.S. Conserved Properties of Hydrogenosomal and Mitochondrial ADP/ATP Carriers: A Common Origin for Both Organelles. EMBO J. 2002, 21, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, S.; Stölting, J.; Zimorski, V.; Rada, P.; Tachezy, J.; Martin, W.F.; Gould, S.B. Conservation of Transit Peptide-Independent Protein Import into the Mitochondrial and Hydrogenosomal Matrix. Genome Biol. Evol. 2015, 7, 2716–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukasawa, Y.; Oda, T.; Tomii, K.; Imai, K. Origin and Evolutionary Alteration of the Mitochondrial Import System in Eukaryotic Lineages. Mol. Biol. Evol. 2017, 34, 1574–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.; Schatz, G. Sequences from a Prokaryotic Genome or the Mouse Dihydrofolate Reductase Gene Can Restore the Import of a Truncated Precursor Protein into Yeast Mitochondria. Proc. Natl. Acad. Sci. USA 1987, 84, 3117–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, C.A.; Preuss, D.; Grisafi, P.; Botstein, D. Many Random Sequences Functionally Replace the Secretion Signal Sequence of Yeast Invertase. Science 1987, 235, 312–317. [Google Scholar] [CrossRef]
- Lemire, B.D.; Fankhauser, C.; Baker, A.; Schatz, G. The Mitochondrial Targeting Function of Randomly Generated Peptide Sequences Correlates with Predicted Helical Amphiphilicity. J. Biol. Chem. 1989, 264, 20206–20215. [Google Scholar]
- Dunn, C.D.; Paavilainen, V.O. Wherever I May Roam: Organellar Protein Targeting and Evolvability. Curr. Opin. Genet. Dev. 2019, 58, 9–16. [Google Scholar] [CrossRef]
- Palmieri, F. The Mitochondrial Transporter Family SLC25: Identification, Properties and Physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Saraste, M.; Walker, J.E. Internal Sequence Repeats and the Path of Polypeptide in Mitochondrial ADP/ATP Translocase. FEBS Lett. 1982, 144, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Forrest, L.R. Structural Symmetry in Membrane Proteins. Annu. Rev. Biophys. 2015, 44, 311–337. [Google Scholar] [CrossRef] [Green Version]
- Runswick, M.J.; Powell, S.J.; Nyren, P.; Walker, J.E. Sequence of the Bovine Mitochondrial Phosphate Carrier Protein: Structural Relationship to ADP/ATP Translocase and the Brown Fat Mitochondria Uncoupling Protein. EMBO J. 1987, 6, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Runswick, M.J.; Walker, J.E.; Bisaccia, F.; Iacobazzi, V.; Palmieri, F. Sequence of the Bovine 2-Oxoglutarate/Malate Carrier Protein: Structural Relationship to Other Mitochondrial Transport Proteins. Biochemistry 1990, 29, 11033–11040. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. The Mitochondrial Carnitine Carrier Protein: cDNA Cloning, Primary Structure and Comparison with Other Mitochondrial Transport Proteins. Biochem. J. 1997, 321 (Pt 3), 713–719. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. Mitochondrial Carrier Proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.E. The Mitochondrial Transporter Family. Curr. Opin. Struct. Biol. 1992, 2, 519–526. [Google Scholar] [CrossRef]
- Nelson, D.R.; Felix, C.M.; Swanson, J.M. Highly Conserved Charge-Pair Networks in the Mitochondrial Carrier Family. J. Mol. Biol. 1998, 277, 285–308. [Google Scholar] [CrossRef]
- Falconi, M.; Chillemi, G.; Di Marino, D.; D’Annessa, I.; Morozzo della Rocca, B.; Palmieri, L.; Desideri, A. Structural Dynamics of the Mitochondrial ADP/ATP Carrier Revealed by Molecular Dynamics Simulation Studies. Proteins 2006, 65, 681–691. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Indiveri, C.; Palmieri, F. Conformation-Dependent Accessibility of Cys-136 and Cys-155 of the Mitochondrial Rat Carnitine/Acylcarnitine Carrier to Membrane-Impermeable SH Reagents. Biochim. Biophys. Acta 2007, 1767, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Cappello, A.R.; Miniero, D.V.; Curcio, R.; Ludovico, A.; Daddabbo, L.; Stipani, I.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Functional and Structural Role of Amino Acid Residues in the Odd-Numbered Transmembrane Alpha-Helices of the Bovine Mitochondrial Oxoglutarate Carrier. J. Mol. Biol. 2007, 369, 400–412. [Google Scholar] [CrossRef]
- Palmieri, F. Diseases Caused by Defects of Mitochondrial Carriers: A Review. Biochim. Biophys. Acta 2008, 1777, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Lauria, G.; Sanchez, P.; Della Rocca, B.M.; Pierri, C.L.; Polizio, F.; Stipani, I.; Desideri, A. Structural-Dynamical Properties of the Transmembrane Segment VI of the Mitochondrial Oxoglutarate Carrier Studied by Site Directed Spin-Labeling. Mol. Membr. Biol. 2008, 25, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Overy, C.; Kunji, E.R.S. The Mechanism of Transport by Mitochondrial Carriers Based on Analysis of Symmetry. Proc. Natl. Acad. Sci. USA 2008, 105, 17766–17771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.M.; Brandolin, G. Structure of Mitochondrial ADP/ATP Carrier in Complex with Carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R.S. Structures of Yeast Mitochondrial ADP/ATP Carriers Support a Domain-Based Alternating-Access Transport Mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Cappello, A.R.; Curcio, R.; Valeria Miniero, D.; Stipani, I.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Functional and Structural Role of Amino Acid Residues in the Even-Numbered Transmembrane Alpha-Helices of the Bovine Mitochondrial Oxoglutarate Carrier. J. Mol. Biol. 2006, 363, 51–62. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R. Structural Changes in the Transport Cycle of the Mitochondrial ADP/ATP Carrier. Curr. Opin. Struct. Biol. 2019, 57, 135–144. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435.e15–447.e15. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [Green Version]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-Nucleotide Evolution Quantifies the Importance of Each Site along the Structure of Mitochondrial Carriers. Cell. Mol. Life Sci. 2014, 71, 349–364. [Google Scholar] [CrossRef]
- Brix, J.; Rüdiger, S.; Bukau, B.; Schneider-Mergener, J.; Pfanner, N. Distribution of Binding Sequences for the Mitochondrial Import Receptors Tom20, Tom22, and Tom70 in a Presequence-Carrying Preprotein and a Non-Cleavable Preprotein. J. Biol. Chem. 1999, 274, 16522–16530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreimendahl, S.; Schwichtenberg, J.; Günnewig, K.; Brandherm, L.; Rassow, J. The Selectivity Filter of the Mitochondrial Protein Import Machinery. BMC Biol. 2020, 18, 156. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N.; Ryan, M.T. The Three Modules of ADP/ATP Carrier Cooperate in Receptor Recruitment and Translocation into Mitochondria. EMBO J. 2001, 20, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, R.S.; Mayor, J.A.; Wood, D.O. The Mitochondrial Tricarboxylate Transport Protein. cDNA Cloning, Primary Structure, and Comparison with Other Mitochondrial Transport Proteins. J. Biol. Chem. 1993, 268, 13682–13690. [Google Scholar]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.C. Identification and Functional Expression of the Mitochondrial Pyruvate Carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef]
- Tavoulari, S.; Thangaratnarajah, C.; Mavridou, V.; Harbour, M.E.; Martinou, J.C.; Kunji, E.R. The Yeast Mitochondrial Pyruvate Carrier Is a Hetero-Dimer in Its Functional State. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Vanderperre, B.; Cermakova, K.; Escoffier, J.; Kaba, M.; Bender, T.; Nef, S.; Martinou, J.C. MPC1-like Is a Placental Mammal-Specific Mitochondrial Pyruvate Carrier Subunit Expressed in Postmeiotic Male Germ Cells. J. Biol. Chem. 2016, 291, 16448–16461. [Google Scholar] [CrossRef] [Green Version]
- Bender, T.; Pena, G.; Martinou, J.C. Regulation of Mitochondrial Pyruvate Uptake by Alternative Pyruvate Carrier Complexes. EMBO J. 2015, 34, 911–924. [Google Scholar] [CrossRef]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar Transporters for Intercellular Exchange and Nutrition of Pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Q. SWEET Sugar Transporters for Phloem Transport and Pathogen Nutrition. New Phytol. 2014, 201, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tao, Y.; Cheung, L.S.; Fan, C.; Chen, L.Q.; Xu, S.; Perry, K.; Frommer, W.B.; Feng, L. Structures of Bacterial Homologues of SWEET Transporters in Two Distinct Conformations. Nature 2014, 515, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Medrano-Soto, A.; Ghazi, F.; Hendargo, K.J.; Moreno-Hagelsieb, G.; Myers, S.; Saier, M.H. Expansion of the Transporter-Opsin-G Protein-Coupled Receptor Superfamily with Five New Protein Families. PLoS ONE 2020, 15, e0231085. [Google Scholar] [CrossRef] [Green Version]
- Xuan, Y.H.; Hu, Y.B.; Chen, L.Q.; Sosso, D.; Ducat, D.C.; Hou, B.H.; Frommer, W.B. Functional Role of Oligomerization for Bacterial and Plant SWEET Sugar Transporter Family. Proc. Natl. Acad. Sci. USA 2013, 110, E3685–E3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yan, C.; Li, Y.; Hirata, K.; Yamamoto, M.; Yan, N.; Hu, Q. Crystal Structure of a Bacterial Homologue of SWEET Transporters. Cell Res. 2014, 24, 1486–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaehme, M.; Guskov, A.; Slotboom, D.J. Crystal Structure of the Vitamin B3 Transporter PnuC, a Full-Length SWEET Homolog. Nat. Struct. Mol. Biol. 2014, 21, 1013–1015. [Google Scholar] [CrossRef]
- Lee, Y.; Nishizawa, T.; Yamashita, K.; Ishitani, R.; Nureki, O. Structural Basis for the Facilitative Diffusion Mechanism by SemiSWEET Transporter. Nat. Commun. 2015, 6, 6112. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhu, Y.; Liu, M.; Zhou, Y.; Lu, G.; Lan, L.; Wang, X.; Zhao, Y.; Zhang, X.C. Molecular Mechanism of Substrate Recognition and Transport by the AtSWEET13 Sugar Transporter. Proc. Natl. Acad. Sci. USA 2017, 114, 10089–10094. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Fastman, N.M.; Venkatakrishnan, A.J.; Frommer, W.B.; Dror, R.O.; Feng, L. Mechanism of Substrate Translocation in an Alternating Access Transporter. Cell 2017, 169, 96.e12–107.e12. [Google Scholar] [CrossRef] [Green Version]
- Brivet, M.; Garcia-Cazorla, A.; Lyonnet, S.; Dumez, Y.; Nassogne, M.C.; Slama, A.; Boutron, A.; Touati, G.; Legrand, A.; Saudubray, J.M. Impaired Mitochondrial Pyruvate Importation in a Patient and a Fetus at Risk. Mol. Genet. Metab. 2003, 78, 186–192. [Google Scholar] [CrossRef]
- Oonthonpan, L.; Rauckhorst, A.J.; Gray, L.R.; Boutron, A.C.; Taylor, E.B. Two Human Patient Mitochondrial Pyruvate Carrier Mutations Reveal Distinct Molecular Mechanisms of Dysfunction. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Clements, J.; Finn, R.D. Skylign: A Tool for Creating Informative, Interactive Logos Representing Sequence Alignments and Profile Hidden Markov Models. BMC Bioinform. 2014, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Tusnády, G.E.; Simon, I. The HMMTOP Transmembrane Topology Prediction Server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Viklund, H.; Bernsel, A.; Skwark, M.; Elofsson, A. SPOCTOPUS: A Combined Predictor of Signal Peptides and Membrane Protein Topology. Bioinformatics 2008, 24, 2928–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, T.; Jones, D.T. Detecting Pore-Lining Regions in Transmembrane Protein Sequences. BMC Bioinform. 2012, 13, 169. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, M.D.; Campagna, D.R.; Haslett, J.N.; Trenor, C.C.; Andrews, N.C. A Mutation in a Mitochondrial Transmembrane Protein Is Responsible for the Pleiotropic Hematological and Skeletal Phenotype of Flexed-Tail (f/f) Mice. Genes Dev. 2001, 15, 652–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kory, N.; Wyant, G.A.; Prakash, G.; Uit de Bos, J.; Bottanelli, F.; Pacold, M.E.; Chan, S.H.; Lewis, C.A.; Wang, T.; Keys, H.R.; et al. SFXN1 Is a Mitochondrial Serine Transporter Required for One-Carbon Metabolism. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Acoba, M.G.; Alpergin, E.S.S.; Renuse, S.; Fernández-del-Río, L.; Lu, Y.W.; Clarke, C.F.; Pandey, A.; Wolfgang, M.J.; Claypool, S.M. The Mitochondrial Carrier SFXN1 Is Critical for Complex III Integrity and Cellular Metabolism. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jackson, T.D.; Hock, D.; Palmer, C.S.; Kang, Y.; Fujihara, K.M.; Clemons, N.J.; Thorburn, D.R.; Stroud, D.A.; Stojanovski, D. The TIM22 Complex Regulates Mitochondrial One-Carbon Metabolism by Mediating the Import of Sideroflexins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Horten, P.; Colina-Tenorio, L.; Rampelt, H. Biogenesis of Mitochondrial Metabolite Carriers. Biomolecules 2020, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal Structure of a Bacterial Homologue of Na+/Cl—Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Boudker, O.; Ryan, R.M.; Yernool, D.; Shimamoto, K.; Gouaux, E. Coupling Substrate and Ion Binding to Extracellular Gate of a Sodium-Dependent Aspartate Transporter. Nature 2007, 445, 387–393. [Google Scholar] [CrossRef]
- Hildick-Smith, G.J.; Cooney, J.D.; Garone, C.; Kremer, L.S.; Haack, T.B.; Thon, J.N.; Miyata, N.; Lieber, D.S.; Calvo, S.E.; Akman, H.O.; et al. Macrocytic Anemia and Mitochondriopathy Resulting from a Defect in Sideroflexin 4. Am. J. Hum. Genet. 2013, 93, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Nowikovsky, K.; Froschauer, E.M.; Zsurka, G.; Samaj, J.; Reipert, S.; Kolisek, M.; Wiesenberger, G.; Schweyen, R.J. The LETM1/YOL027 Gene Family Encodes a Factor of the Mitochondrial K+ Homeostasis with a Potential Role in the Wolf-Hirschhorn Syndrome. J. Biol. Chem. 2004, 279, 30307–30315. [Google Scholar] [CrossRef] [Green Version]
- Froschauer, E.; Nowikovsky, K.; Schweyen, R.J. Electroneutral K+/H+ Exchange in Mitochondrial Membrane Vesicles Involves Yol027/Letm1 Proteins. Biochim. Biophys. Acta 2005, 1711, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-Wide RNAi Screen Identifies Letm1 as a Mitochondrial Ca2+/H+ Antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional Reconstitution of the Mitochondrial Ca2+/H+ Antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Fu, Z.; Ji, Y.; Guan, X.; Guo, S.; Ding, Z.; Yang, X.; Cong, Y.; Shen, Y. Leucine Zipper-EF-Hand Containing Transmembrane Protein 1 (LETM1) Forms a Ca2+/H+ Antiporter. Sci. Rep. 2016, 6, 34174. [Google Scholar] [CrossRef] [Green Version]
- Nowikovsky, K.; Bernardi, P. LETM1 in Mitochondrial Cation Transport. Front. Physiol. 2014, 5, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordes, F.S.; Bright, J.N.; Sansom, M.S.P. Proline-Induced Distortions of Transmembrane Helices. J. Mol. Biol. 2002, 323, 951–960. [Google Scholar] [CrossRef]
- Law, E.C.; Wilman, H.R.; Kelm, S.; Shi, J.; Deane, C.M. Examining the Conservation of Kinks in Alpha Helices. PLoS ONE 2016, 11, e0157553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting Sequence Signals in Targeting Peptides Using Deep Learning. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csere, P.; Lill, R.; Kispal, G. Identification of a Human Mitochondrial ABC Transporter, the Functional Orthologue of Yeast Atm1p. FEBS Lett. 1998, 441, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Hogue, D.L.; Liu, L.; Ling, V. Identification and Characterization of a Mammalian Mitochondrial ATP-Binding Cassette Membrane Protein. J. Mol. Biol. 1999, 285, 379–389. [Google Scholar] [CrossRef]
- Zhang, F.; Hogue, D.L.; Liu, L.; Fisher, C.L.; Hui, D.; Childs, S.; Ling, V. M-ABC2, a New Human Mitochondrial ATP-Binding Cassette Membrane Protein. FEBS Lett. 2000, 478, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.C.; Du, G.; Fukuda, Y.; Sun, D.; Sampath, J.; Mercer, K.E.; Wang, J.; Sosa-Pineda, B.; Murti, K.G.; Schuetz, J.D. Identification of a Mammalian Mitochondrial Porphyrin Transporter. Nature 2006, 443, 586–589. [Google Scholar] [CrossRef]
- Kiss, K.; Brozik, A.; Kucsma, N.; Toth, A.; Gera, M.; Berry, L.; Vallentin, A.; Vial, H.; Vidal, M.; Szakacs, G. Shifting the Paradigm: The Putative Mitochondrial Protein ABCB6 Resides in the Lysosomes of Cells and in the Plasma Membrane of Erythrocytes. PLoS ONE 2012, 7, e37378. [Google Scholar] [CrossRef] [Green Version]
- Kiss, K.; Kucsma, N.; Brozik, A.; Tusnady, G.E.; Bergam, P.; van Niel, G.; Szakacs, G. Role of the N-Terminal Transmembrane Domain in the Endo-Lysosomal Targeting and Function of the Human ABCB6 Protein. Biochem. J. 2015, 467, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Graf, S.A.; Haigh, S.E.; Corson, E.D.; Shirihai, O.S. Targeting, Import, and Dimerization of a Mammalian Mitochondrial ATP Binding Cassette (ABC) Transporter, ABCB10 (ABC-Me). J. Biol. Chem. 2004, 279, 42954–42963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Allikmets, R.; Gerrard, B.; Stewart, C.; Kistler, A.; Shafer, B.; Michaelis, S.; Strathern, J. Mapping and Sequencing of Two Yeast Genes Belonging to the ATP-Binding Cassette Superfamily. Yeast 1994, 10, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Stiller, S.B.; Höpker, J.; Oeljeklaus, S.; Schütze, C.; Schrempp, S.G.; Vent-Schmidt, J.; Horvath, S.E.; Frazier, A.E.; Gebert, N.; van der Laan, M.; et al. Mitochondrial OXA Translocase Plays a Major Role in Biogenesis of Inner-Membrane Proteins. Cell Metab. 2016, 23, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintre, C.A.; Pike, A.C.W.; Li, Q.; Kim, J.I.; Barr, A.J.; Goubin, S.; Shrestha, L.; Yang, J.; Berridge, G.; Ross, J.; et al. Structures of ABCB10, a Human ATP-Binding Cassette Transporter in Apo- and Nucleotide-Bound States. Proc. Natl. Acad. Sci. USA 2013, 110, 9710–9715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, V.; Pierik, A.J.; Lill, R. Crystal Structures of Nucleotide-Free and Glutathione-Bound Mitochondrial ABC Transporter Atm1. Science 2014, 343, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 Transporter: Gene Structure and Mutation Causing X-Linked Sideroblastic Anemia with Ataxia with Disruption of Cytosolic Iron-Sulfur Protein Maturation. Blood 2000, 96, 3256–3264. [Google Scholar] [CrossRef]
- Seguin, A.; Takahashi-Makise, N.; Yien, Y.Y.; Huston, N.C.; Whitman, J.C.; Musso, G.; Wallace, J.A.; Bradley, T.; Bergonia, H.A.; Kafina, M.D.; et al. Reductions in the Mitochondrial ABC Transporter Abcb10 Affect the Transcriptional Profile of Heme Biosynthesis Genes. J. Biol. Chem. 2017, 292, 16284–16299. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX Is an Essential Component of Mitochondrial Na+/Ca2+ Exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Khananshvili, D. Distinction between the Two Basic Mechanisms of Cation Transport in the Cardiac Na+-Ca2+ Exchange System. Biochemistry 1990, 29, 2437–2442. [Google Scholar] [CrossRef]
- Pitts, B.J. Stoichiometry of Sodium-Calcium Exchange in Cardiac Sarcolemmal Vesicles. Coupling to the Sodium Pump. J. Biol. Chem. 1979, 254, 6232–6235. [Google Scholar]
- Reeves, J.P.; Hale, C.C. The Stoichiometry of the Cardiac Sodium-Calcium Exchange System. J. Biol. Chem. 1984, 259, 7733–7739. [Google Scholar] [PubMed]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-Calcium Exchange Is Mediated by a Distinct Potassium-Independent Sodium-Calcium Exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernansanz-Agustín, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Piña, T.; Moreno, L.; Izquierdo-Álvarez, A.; Cabrera-García, J.D.; Cortés, A.; et al. Na+ Controls Hypoxic Signalling by the Mitochondrial Respiratory Chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Refaeli, B.; Giladi, M.; Hiller, R.; Khananshvili, D. Structure-Based Engineering of Lithium-Transport Capacity in an Archaeal Sodium-Calcium Exchanger. Biochemistry 2016, 55, 1673–1676. [Google Scholar] [CrossRef]
- Roy, S.; Dey, K.; Hershfinkel, M.; Ohana, E.; Sekler, I. Identification of Residues That Control Li+ versus Na+ Dependent Ca2+ Exchange at the Transport Site of the Mitochondrial NCLX. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 997–1008. [Google Scholar] [CrossRef]
- Liao, J.; Li, H.; Zeng, W.; Sauer, D.B.; Belmares, R.; Jiang, Y. Structural Insight into the Ion-Exchange Mechanism of the Sodium/Calcium Exchanger. Science 2012, 335, 686–690. [Google Scholar] [CrossRef]
- Liao, J.; Marinelli, F.; Lee, C.; Huang, Y.; Faraldo-Gómez, J.D.; Jiang, Y. Mechanism of Extracellular Ion Exchange and Binding-Site Occlusion in a Sodium/Calcium Exchanger. Nat. Struct. Mol. Biol. 2016, 23, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, T.; Kita, S.; Maturana, A.D.; Furuya, N.; Hirata, K.; Kasuya, G.; Ogasawara, S.; Dohmae, N.; Iwamoto, T.; Ishitani, R.; et al. Structural Basis for the Counter-Transport Mechanism of a H2+/Ca2+ Exchanger. Science 2013, 341, 168–172. [Google Scholar] [CrossRef]
- Battaglino, R.A.; Pham, L.; Morse, L.R.; Vokes, M.; Sharma, A.; Odgren, P.R.; Yang, M.; Sasaki, H.; Stashenko, P. NHA-Oc/NHA2: A Mitochondrial Cation-Proton Antiporter Selectively Expressed in Osteoclasts. Bone 2008, 42, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Fuster, D.G.; Zhang, J.; Shi, M.; Bobulescu, I.A.; Andersson, S.; Moe, O.W. Characterization of the Sodium/Hydrogen Exchanger NHA2. J. Am. Soc. Nephrol. 2008, 19, 1547–1556. [Google Scholar] [CrossRef] [Green Version]
- Donowitz, M.; Ming Tse, C.; Fuster, D. SLC9/NHE Gene Family, a Plasma Membrane and Organellar Family of Na+/H+ Exchangers. Mol. Asp. Med. 2013, 34, 236–251. [Google Scholar] [CrossRef] [Green Version]
- Masrati, G.; Dwivedi, M.; Rimon, A.; Gluck-Margolin, Y.; Kessel, A.; Ashkenazy, H.; Mayrose, I.; Padan, E.; Ben-Tal, N. Broad Phylogenetic Analysis of Cation/Proton Antiporters Reveals Transport Determinants. Nat. Commun. 2018, 9, 4205. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Hiller, K.; Grote, A.; Scheer, M.; Münch, R.; Jahn, D. PrediSi: Prediction of Signal Peptides and Their Cleavage Positions. Nucleic Acids Res. 2004, 32, W375–W379. [Google Scholar] [CrossRef]
- Console, L.; Scalise, M.; Tarmakova, Z.; Coe, I.R.; Indiveri, C. N-Linked Glycosylation of Human SLC1A5 (ASCT2) Transporter Is Critical for Trafficking to Membrane. Biochim. Biophys. Acta 2015, 1853, 1636–1645. [Google Scholar] [CrossRef] [Green Version]
- Canul-Tec, J.C.; Assal, R.; Cirri, E.; Legrand, P.; Brier, S.; Chamot-Rooke, J.; Reyes, N. Structure and Allosteric Inhibition of Excitatory Amino Acid Transporter 1. Nature 2017, 544, 446–451. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino Acid Transporters Revisited: New Views in Health and Disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Welbourne, T.; Routh, R.; Yudkoff, M.; Nissim, I. The Glutamine/Glutamate Couplet and Cellular Function. News Physiol. Sci. 2001, 16, 157–160. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyimesi, G.; Hediger, M.A. Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules 2020, 10, 1611. https://doi.org/10.3390/biom10121611
Gyimesi G, Hediger MA. Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules. 2020; 10(12):1611. https://doi.org/10.3390/biom10121611
Chicago/Turabian StyleGyimesi, Gergely, and Matthias A. Hediger. 2020. "Sequence Features of Mitochondrial Transporter Protein Families" Biomolecules 10, no. 12: 1611. https://doi.org/10.3390/biom10121611
APA StyleGyimesi, G., & Hediger, M. A. (2020). Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules, 10(12), 1611. https://doi.org/10.3390/biom10121611