Bioinformatics Analysis of Global Proteomic and Phosphoproteomic Data Sets Revealed Activation of NEK2 and AURKA in Cancers


Abstract
:1. Introduction
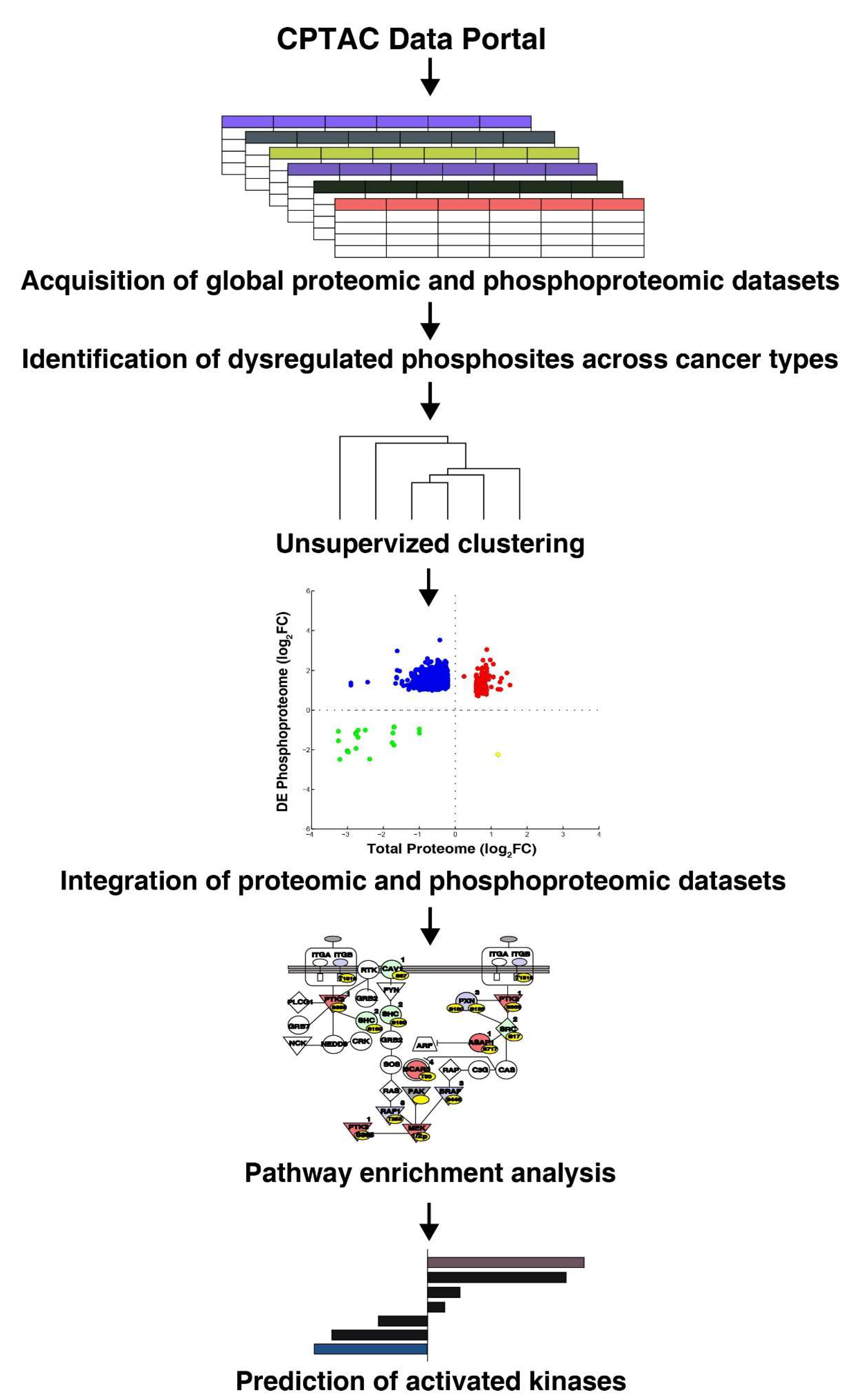
2. Materials and Methods
2.1. Phosphoproteomic and Proteomic Data Mining
2.2. Clustering of Phosphoproteomic Data Sets
2.3. Kinome Map
2.4. Pathway Analysis
2.5. Protein–Protein Interaction Network Analysis
2.6. Quadrant Plot for Comparative Expression and Phosphorylation Levels of Proteins
2.7. Prediction of Activated Kinases Using Kinase-Substrate Enrichment Analysis (KSEA) Tool and Overall Survival Estimates
2.8. Motif Analysis
3. Results
3.1. Dysregulation of Protein Phosphorylation in Cancer Types
3.2. Epithelial-Mesenchymal Transition (EMT) and Its Molecular Regulation Across Six Cancer Types
3.3. A Common Phosphorylation Signature Identified
3.4. Unique Phosphorylation of Proteins Identified Through the Integration of Global Protein Expression of the Phosphopeptide Signature
3.5. Hyperphosphorylated Kinases with Basal Level Expression Were Identified
3.6. Proline-Directed Motifs Were Highly Phosphorylated Across Five Cancer Types
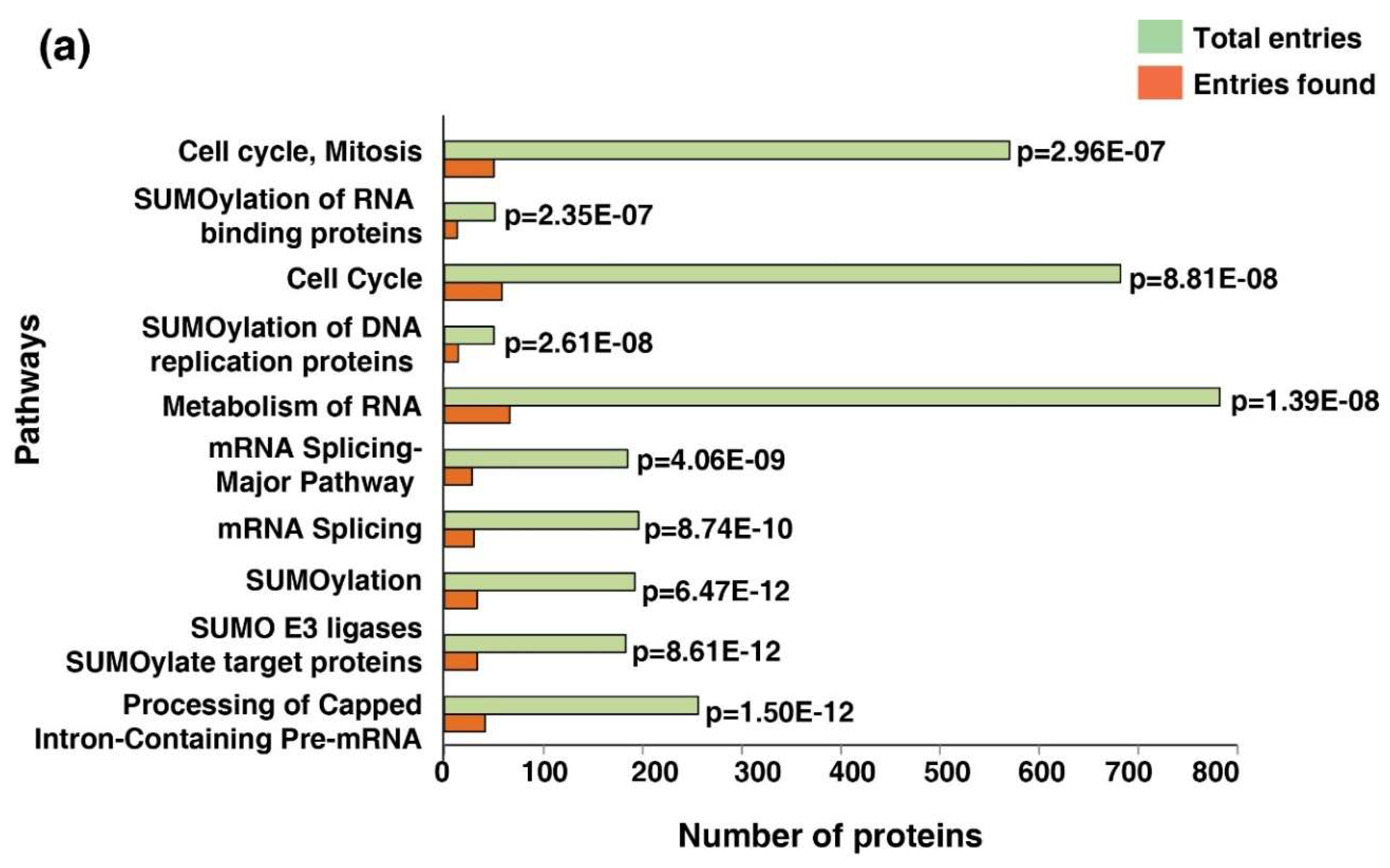
3.7. Cell Cycle Pathway Was Enriched Across the Five Cancer Types
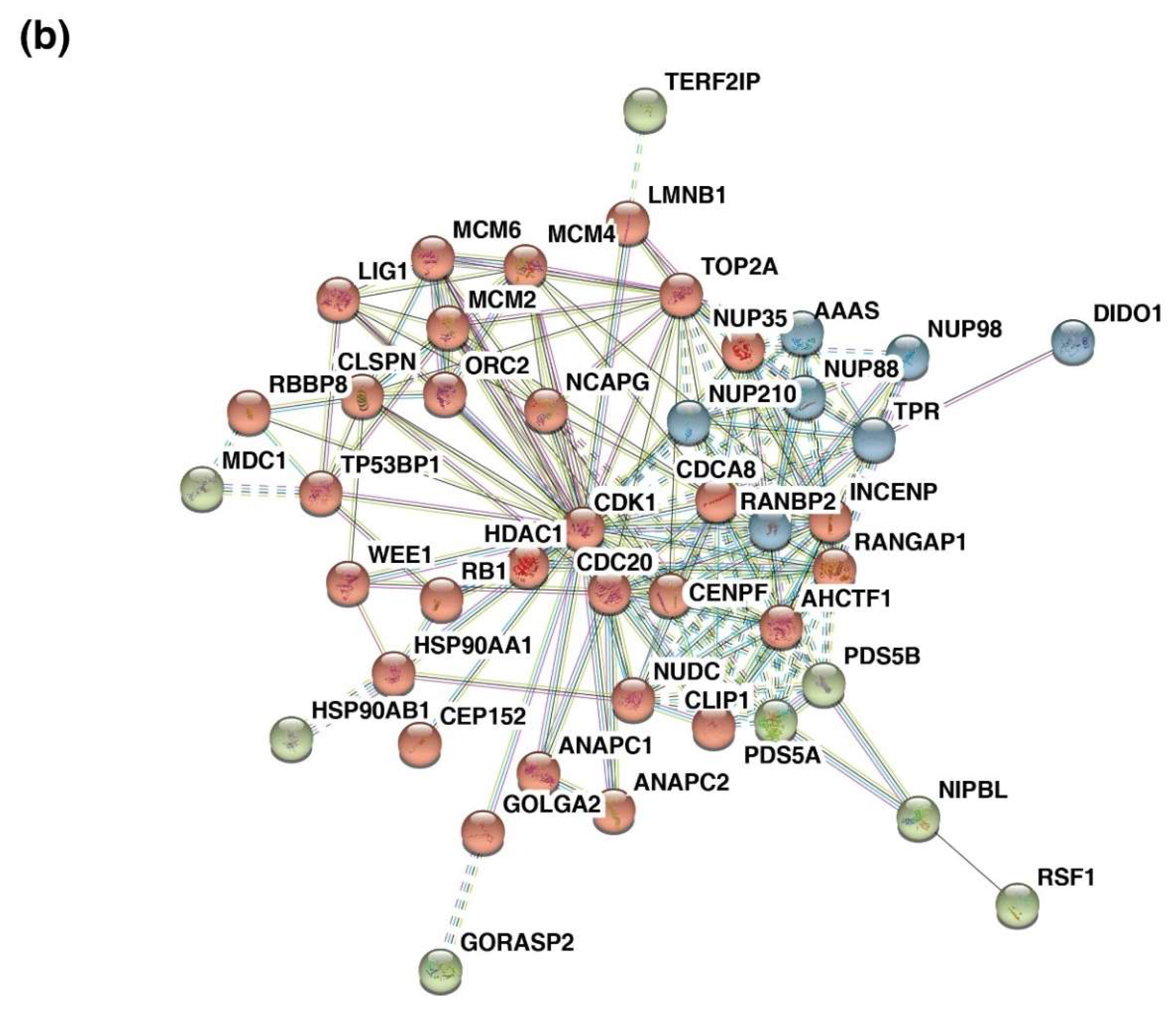
3.8. Protein Interaction Clusters Common across Five Cancers
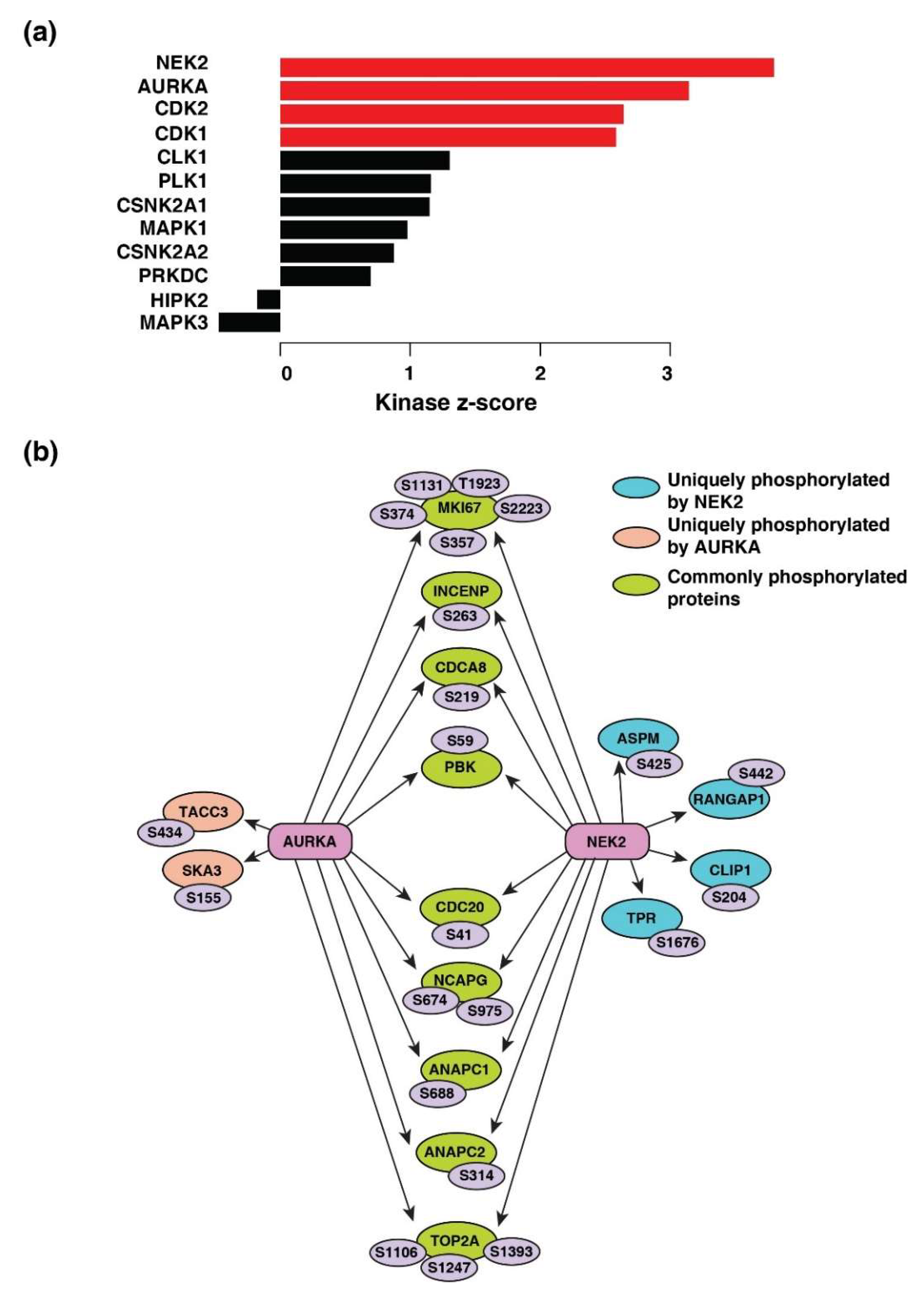
3.9. Serine/Threonine-Protein Kinase Nek2 (NEK2) and Aurora Kinase A (AURKA) Are the Most Predicted Activated Kinases across the Five Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef]
- Deb, B.; George, I.A.; Sharma, J.; Kumar, P. Phosphoproteomics Profiling to Identify Altered Signaling Pathways and Kinase-Targeted Cancer Therapies. Methods Mol. Biol. 2020, 2051, 241–264. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Dazert, E.; Colombi, M.; Boldanova, T.; Moes, S.; Adametz, D.; Quagliata, L.; Roth, V.; Terracciano, L.; Heim, M.H.; Jenoe, P.; et al. Quantitative proteomics and phosphoproteomics on serial tumor biopsies from a sorafenib-treated HCC patient. Proc. Natl. Acad. Sci. USA 2016, 113, 1381–1386. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Deb, B.; Puttamallesh, V.N.; Gondkar, K.; Thiery, J.P.; Gowda, H.; Kumar, P. Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.V.; Nayakanti, D.S.; Sathe, G.; George, I.A.; Gowda, H.; Kumar, P. Targeted Proteomics as a Tool for Quantifying Urine-Based Biomarkers. Methods Mol. Biol. 2020, 2051, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef] [PubMed]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef]
- Cicenas, J.; Valius, M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers (Basel) 2014, 6, 2224–2242. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Cicenas, E. Multi-kinase inhibitors, AURKs and cancer. Med. Oncol. 2016, 33, 43. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000 Res. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Aasen, S.N.; Parajuli, H.; Hoang, T.; Feng, Z.; Stokke, K.; Wang, J.; Roy, K.; Bjerkvig, R.; Knappskog, S.; Thorsen, F. Effective Treatment of Metastatic Melanoma by Combining MAPK and PI3K Signaling Pathway Inhibitors. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Staderini, F.; Cianchi, F.; Badii, B.; Skalamera, I.; Fiorenza, G.; Foppa, C.; Qirici, E.; Perigli, G. A unique presentation of a renal clear cell carcinoma with atypical metastases. Int. J. Surg. Case Rep. 2015, 11, 29–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landolt, L.; Eikrem, O.; Strauss, P.; Scherer, A.; Lovett, D.H.; Beisland, C.; Finne, K.; Osman, T.; Ibrahim, M.M.; Gausdal, G.; et al. Clear Cell Renal Cell Carcinoma is linked to Epithelial-to-Mesenchymal Transition and to Fibrosis. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thu, K.L.; Soria-Bretones, I.; Mak, T.W.; Cescon, D.W. Targeting the cell cycle in breast cancer: Towards the next phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullany, L.E.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.; Stevens, J.R.; Wolff, R.K.; Slattery, M.L. miRNA involvement in cell cycle regulation in colorectal cancer cases. Genes Cancer 2018, 9, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Baldi, A.; De Luca, A.; Esposito, V.; Campioni, M.; Spugnini, E.P.; Citro, G. Tumor suppressors and cell-cycle proteins in lung cancer. Pathol. Res. Int. 2011, 2011, 5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenzi, B.; Schiavon, G.; Silletta, M.; Santini, D.; Perrone, G.; Di Marino, M.; Angeletti, S.; Baldi, A.; Tonini, G. Cell cycle alterations and lung cancer. Histol. Histopathol. 2006, 21, 423–435. [Google Scholar] [CrossRef]
- Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- D′Andrilli, G.; Kumar, C.; Scambia, G.; Giordano, A. Cell cycle genes in ovarian cancer: Steps toward earlier diagnosis and novel therapies. Clin. Cancer Res. 2004, 10, 8132–8141. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Krek, W.; Nigg, E.A. Cell cycle regulation of vertebrate p34cdc2 activity: Identification of Thr161 as an essential in vivo phosphorylation site. New Biol. 1992, 4, 323–329. [Google Scholar] [PubMed]
- Norbury, C.; Blow, J.; Nurse, P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991, 10, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.F.; Wang, R.; Liang, Q.; Liang, J.; Li, W.; Jung, S.Y.; Qin, J.; Lin, S.H.; Kuang, J. Dissecting the M phase-specific phosphorylation of serine-proline or threonine-proline motifs. Mol. Biol. Cell 2010, 21, 1470–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Rellos, P.; Ivins, F.J.; Baxter, J.E.; Pike, A.; Nott, T.J.; Parkinson, D.M.; Das, S.; Howell, S.; Fedorov, O.; Shen, Q.Y.; et al. Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 2007, 282, 6833–6842. [Google Scholar] [CrossRef] [Green Version]
- Coxon, C.R.; Wong, C.; Bayliss, R.; Boxall, K.; Carr, K.H.; Fry, A.M.; Hardcastle, I.R.; Matheson, C.J.; Newell, D.R.; Sivaprakasam, M.; et al. Structure-guided design of purine-based probes for selective Nek2 inhibition. Oncotarget 2017, 8, 19089–19124. [Google Scholar] [CrossRef]
- Garcia-Aranda, M.; Redondo, M. Protein Kinase Targets in Breast Cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Franqui Machin, R.; Gu, Z.; Zhan, F. Role of NEK2A in human cancer and its therapeutic potentials. Biomed. Res. Int. 2015, 2015, 2461. [Google Scholar] [CrossRef] [Green Version]
- Cirak, Y.; Furuncuoglu, Y.; Yapicier, O.; Aksu, A.; Cubukcu, E. Aurora A overexpression in breast cancer patients induces taxane resistance and results in worse prognosis. J. BUON 2015, 20, 1414–1419. [Google Scholar] [PubMed]
- Do, T.V.; Xiao, F.; Bickel, L.E.; Klein-Szanto, A.J.; Pathak, H.B.; Hua, X.; Howe, C.; O′Brien, S.W.; Maglaty, M.; Ecsedy, J.A.; et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene 2014, 33, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, B.; Nymoen, D.A.; Elgaaen, B.V.; Staff, A.C.; Trope, C.G.; Kaern, J.; Reich, R.; Falkenthal, T.E. BUB1 mRNA is significantly co-expressed with AURKA and AURKB mRNA in advanced-stage ovarian serous carcinoma. Virchows Arch. 2014, 464, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Casorzo, L.; Dell′Aglio, C.; Sarotto, I.; Risio, M. Aurora kinase A gene copy number is associated with the malignant transformation of colorectal adenomas but not with the serrated neoplasia progression. Hum. Pathol. 2015, 46, 411–418. [Google Scholar] [CrossRef]
- Zhang, T.; Fields, J.Z.; Opdenaker, L.; Otevrel, T.; Masuda, E.; Palazzo, J.P.; Isenberg, G.A.; Goldstein, S.D.; Brand, M.; Boman, B.M. Survivin-induced Aurora-B kinase activation: A mechanism by which APC mutations contribute to increased mitoses during colon cancer development. Am. J. Pathol. 2010, 177, 2816–2826. [Google Scholar] [CrossRef]
- Katsha, A.; Belkhiri, A.; Goff, L.; El-Rifai, W. Aurora kinase A in gastrointestinal cancers: Time to target. Mol. Cancer 2015, 14, 106. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yue, C.F.; Zhou, W.H.; Qian, Y.M.; Zhang, Y.; Wang, S.W.; Liu, A.W.; Liu, Q. Aurora-A contributes to cisplatin resistance and lymphatic metastasis in non-small cell lung cancer and predicts poor prognosis. J. Transl. Med. 2014, 12, 200. [Google Scholar] [CrossRef] [Green Version]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Details | CPTAC Cancer Proteome Confirmatory Colon Study | CPTAC Ovarian Cancer Confirmatory Study | CPTAC Breast Cancer Confirmatory Study | CPTAC Uterine Corpus Endometrial Carcinoma (UCEC) Discovery Study | CPTAC Clear Cell Renal Cell Carcinoma (CCRCC) Discovery Study | CPTAC Lung Adenocarcinoma (LUAD) Discovery Study |
|---|---|---|---|---|---|---|
| CPTAC Accession Number | S037 | S038 | S039 | S043 | S044 | S046 |
| Tumor Sample Count | 97 | 84 | 133 | 100 | 110 | 113 |
| Adjacent Normal Sample Count | 100 | 19 | 18 | 40 | 84 | 102 |
| Unique Phosphosites Identified | 40,302 | 43,811 | 65,068 | 43,842 | 41,809 | 45,671 |
| Unique Protein Identified | 4724 | 5299 | 5852 | 6155 | 5740 | 6020 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deb, B.; Sengupta, P.; Sambath, J.; Kumar, P. Bioinformatics Analysis of Global Proteomic and Phosphoproteomic Data Sets Revealed Activation of NEK2 and AURKA in Cancers. Biomolecules 2020, 10, 237. https://doi.org/10.3390/biom10020237
Deb B, Sengupta P, Sambath J, Kumar P. Bioinformatics Analysis of Global Proteomic and Phosphoproteomic Data Sets Revealed Activation of NEK2 and AURKA in Cancers. Biomolecules. 2020; 10(2):237. https://doi.org/10.3390/biom10020237
Chicago/Turabian StyleDeb, Barnali, Pratyay Sengupta, Janani Sambath, and Prashant Kumar. 2020. "Bioinformatics Analysis of Global Proteomic and Phosphoproteomic Data Sets Revealed Activation of NEK2 and AURKA in Cancers" Biomolecules 10, no. 2: 237. https://doi.org/10.3390/biom10020237
APA StyleDeb, B., Sengupta, P., Sambath, J., & Kumar, P. (2020). Bioinformatics Analysis of Global Proteomic and Phosphoproteomic Data Sets Revealed Activation of NEK2 and AURKA in Cancers. Biomolecules, 10(2), 237. https://doi.org/10.3390/biom10020237