From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds


Abstract
:1. Introduction
2. Antitumoral Compounds Originated from Marine Flora
2.1. Bacteria, Actinobacteria, and Cyanobacteria
2.2. Fungi
2.3. Microalgae
2.4. Macroalgae
2.5. Mangroves and Other Higher Plants
3. Antitumoral Compounds Originated from Marine Invertebrate Fauna
3.1. Sponges
3.2. Tunicates
3.3. Mollusks
3.4. Bryozoans
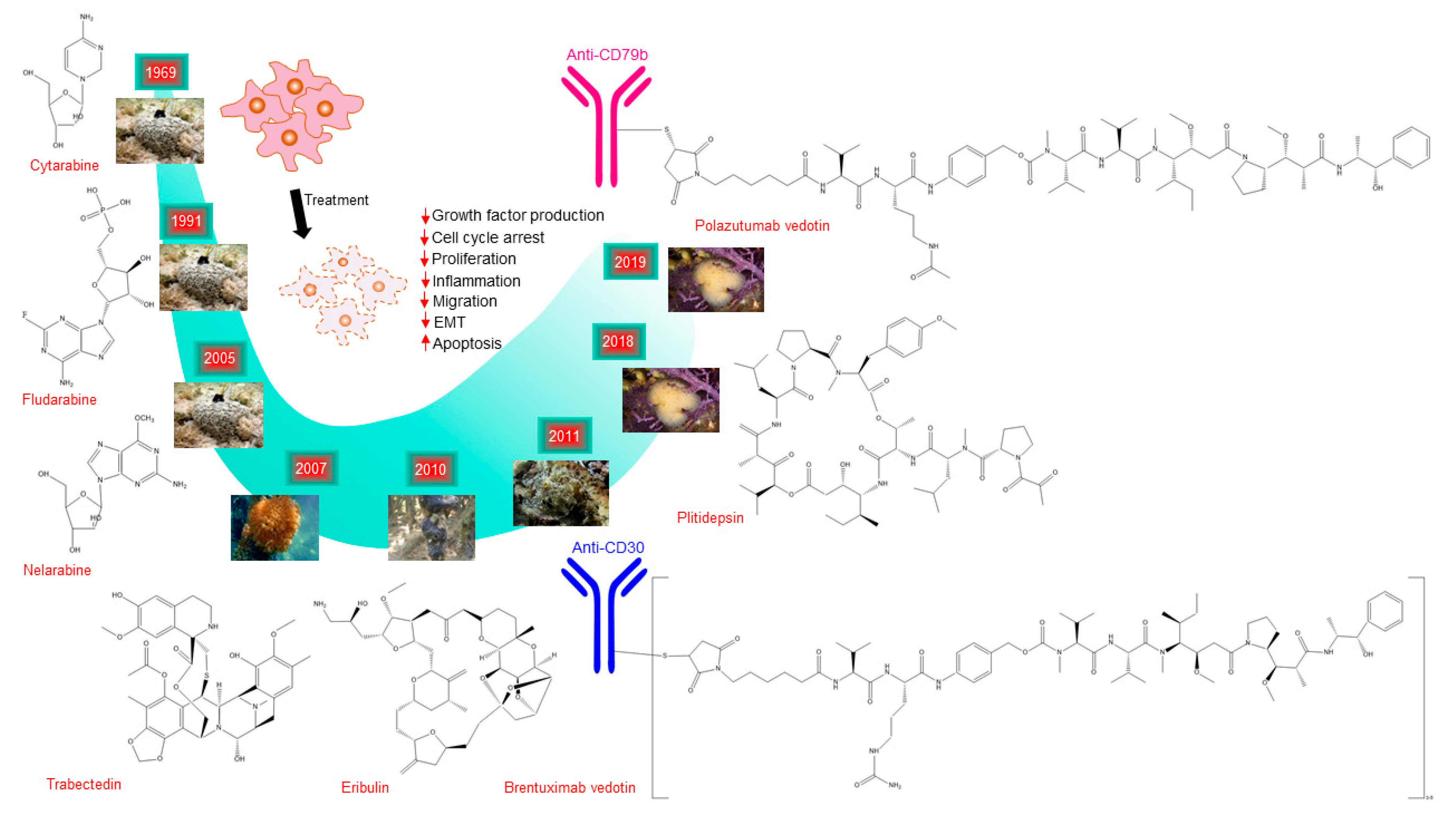
4. Marine Compounds Approved for Cancer Treatment
5. Limitations of Antitumoral Marine Compounds for their Clinical Development and Strategies to Overcome the Limitations
5.1. Lack of Sustainable Supply
5.2. Low Production of Bioactive Compounds
5.3. Poor Technical Infrastructure
5.4. Structural Complexity of the Marine Compounds
5.5. Correct Taxonomic Determination
5.6. Moderate Efficacy
5.7. High Market Value
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Kim, S.K. Marine-Derived Nutraceuticals: Trends and Prospects. Mari. Nutraceuticals 2013. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: a new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Shen, X.; El Sayed, K.A.; Dunbar, D.C.; Perry, T.L.; Wilkins, S.P.; Hamann, M.T.; Bobzin, S.; Huesing, J.; Camp, R.; et al. Marine natural products as prototype agrochemical agents. J. Agric. Food Chem. 2003, 51, 2246–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suleria, H.A.; Osborne, S.; Masci, P.; Gobe, G. Marine-Based Nutraceuticals: An Innovative Trend in the Food and Supplement Industries. Mar. Drugs 2015, 13, 6336–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, M.H.; Blunt, J.W.; Dumdei, E.J.; Hickford, S.J.; Lill, R.E.; Li, S.; Battershill, C.N.; Duckworth, A.R. The discovery and development of marine compounds with pharmaceutical potential. J. Biotechnol. 1999, 70, 15–25. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [Green Version]
- Erwin, P.M.; Lopez-Legentil, S.; Schuhmann, P.W. The pharmaceutical value of marine biodiversity for anti-cancer drug discovery. Ecol. Econ. 2010, 70, 445–451. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R.J. The Isolation of a New Thymine Pentoside from Sponges. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Kremer, W.B. Drugs five years later: cytarabine. Ann. Intern. Med. 1975, 82, 684–688. [Google Scholar] [CrossRef]
- Lowenberg, B.; Pabst, T.; Vellenga, E.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Biemond, B.J.; Gratwohl, A.; et al. Cytarabine dose for acute myeloid leukemia. N. Engl. J. Med. 2011, 364, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathiresan, K.A.D. Current issue of microbiology. ENVIS Centre Newsl. 2005, 4, 3–5. [Google Scholar]
- Sithranga Boopathy, N.; Kathiresan, K. Anticancer Drugs from Marine Flora: An Overview. J. Oncol. 2010, 2010, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Kim, M. Marine natural flora: A potent source of anticancer metabolites. Indian J. Geo Mar. Sci. 2016, 45, 1412–1421. [Google Scholar]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef]
- Mooberry, S.L.; Leal, R.M.; Tinley, T.L.; Luesch, H.; Moore, R.E.; Corbett, T.H. The molecular pharmacology of symplostatin 1: a new antimitotic dolastatin 10 analog. Int. J. Cancer 2003, 104, 512–521. [Google Scholar] [CrossRef]
- Kobayashi, M.; Natsume, T.; Tamaoki, S.; Watanabe, J.; Asano, H.; Mikami, T.; Miyasaka, K.; Miyazaki, K.; Gondo, M.; Sakakibara, K.; et al. Antitumor activity of TZT-1027, a novel dolastatin 10 derivative. Jpn. J. Cancer Res. 1997, 88, 316–327. [Google Scholar] [CrossRef]
- Horti, J.; Juhasz, E.; Monostori, Z.; Maeda, K.; Eckhardt, S.; Bodrogi, I. Phase I study of TZT-1027, a novel synthetic dolastatin 10 derivative, for the treatment of patients with non-small cell lung cancer. Cancer Chemother. Pharmacol. 2008, 62, 173–180. [Google Scholar] [CrossRef]
- Abreu, P.A.; Sousa, T.S.; Jimenez, P.C.; Wilke, D.V.; Rocha, D.D.; Freitas, H.P.; Pessoa, O.D.; La Clair, J.J.; Costa-Lotufo, L.V. Identification of pyrroloformamide as a cytokinesis modulator. Chembiochem 2014, 15, 501–506. [Google Scholar] [CrossRef]
- Guimaraes, L.A.; Jimenez, P.C.; Sousa Tda, S.; Freitas, H.P.; Rocha, D.D.; Wilke, D.V.; Martin, J.; Reyes, F.; Deusdenia Loiola Pessoa, O.; Costa-Lotufo, L.V. Chromomycin A2 induces autophagy in melanoma cells. Mar. Drugs 2014, 12, 5839–5855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa Tda, S.; Jimenez, P.C.; Ferreira, E.G.; Silveira, E.R.; Braz-Filho, R.; Pessoa, O.D.; Costa-Lotufo, L.V. Anthracyclinones from Micromonospora sp. J. Nat. Prod. 2012, 75, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barria, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpinski, T.M.; Adamczak, A. Anticancer Activity of Bacterial Proteins and Peptides. Pharmaceutics 2018, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.Y.; Williams, P.G.; Kwon, H.C.; Jensen, P.R.; Fenical, W. Lucentamycins A-D, cytotoxic peptides from the marine-derived actinomycete Nocardiopsis lucentensis. J. Nat. Prod. 2007, 70, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Hua, H.M.; Pei, Y.H.; Yao, X.S. Three new cytotoxic cyclic acylpeptides from marine Bacillus sp. Chem. Pharm. Bull. 2004, 52, 1029–1030. [Google Scholar] [CrossRef] [Green Version]
- Um, S.; Choi, T.J.; Kim, H.; Kim, B.Y.; Kim, S.H.; Lee, S.K.; Oh, K.B.; Shin, J.; Oh, D.C. Ohmyungsamycins A and B: cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Org. Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef]
- Matsuo, Y.; Kanoh, K.; Imagawa, H.; Adachi, K.; Nishizawa, M.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542. II. Physico-chemical properties and structural elucidation. J. Antibiot. 2007, 60, 256–260. [Google Scholar] [CrossRef]
- Matsuo, Y.; Kanoh, K.; Yamori, T.; Kasai, H.; Katsuta, A.; Adachi, K.; Shin-Ya, K.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542. I. Fermentation, isolation and biological activities. J. Antibiot. 2007, 60, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Marine Fungi: A Source of Potential Anticancer Compounds. Front. Microbiol. 2017, 8, 2536. [Google Scholar] [CrossRef]
- Wahl, M.; Goecke, F.; Labes, A.; Dobretsov, S.; Weinberger, F. The second skin: ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.H.; Wang, C.Y.; Mandi, A.; Li, X.M.; Hu, X.Y.; Kassack, M.U.; Kurtan, T.; Wang, B.G. Three Diketopiperazine Alkaloids with Spirocyclic Skeletons and One Bisthiodiketopiperazine Derivative from the Mangrove-Derived Endophytic Fungus Penicillium brocae MA-231. Org. Lett. 2016, 18, 5304–5307. [Google Scholar] [CrossRef]
- Zheng, C.C.Y.; Jiang, L.; Shi, X.M. Antiproliferative metabolites from the endophytic fungus Penicillium sp. FJ-1 isolated from a mangrove Avicennia marina. Phytochem. Lett. 2014, 10, 273–275. [Google Scholar] [CrossRef]
- Mita, M.M.; Spear, M.A.; Yee, L.K.; Mita, A.C.; Heath, E.I.; Papadopoulos, K.P.; Federico, K.C.; Reich, S.D.; Romero, O.; Malburg, L.; et al. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin(NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892–5899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millward, M.; Mainwaring, P.; Mita, A.; Federico, K.; Lloyd, G.K.; Reddinger, N.; Nawrocki, S.; Mita, M.; Spear, M.A. Phase 1 study of the novel vascular disrupting agent plinabulin (NPI-2358) and docetaxel. Invest. New Drugs 2012, 30, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-Hack, M.E.; Abdelnour, S.; Alagawany, M.; Abdo, M.; Sakr, M.A.; Khafaga, A.F.; Mahgoub, S.A.; Elnesr, S.S.; Gebriel, M.G. Microalgae in modern cancer therapy: Current knowledge. Biomed. Pharmacother. 2019, 111, 42–50. [Google Scholar] [CrossRef]
- Martinez Andrade, K.A.; Lauritano, C.; Romano, G.; Ianora, A. Marine Microalgae with Anti-Cancer Properties. Mar. Drugs 2018, 16, 165. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, P.N.; Ashokkumar, P.; Sudhandiran, G. Antioxidative and antiproliferative effects of astaxanthin during the initiation stages of 1,2-dimethyl hydrazine-induced experimental colon carcinogenesis. Fundam Clin. Pharmacol. 2009, 23, 225–234. [Google Scholar] [CrossRef]
- Lin, P.Y.; Tsai, C.T.; Chuang, W.L.; Chao, Y.H.; Pan, I.H.; Chen, Y.K.; Lin, C.C.; Wang, B.Y. Chlorella sorokiniana induces mitochondrial-mediated apoptosis in human non-small cell lung cancer cells and inhibits xenograft tumor growth in vivo. BMC Complement. Altern. Med. 2017, 17, 88. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Thomas, N.V.; Li, X. Anticancer compounds from marine macroalgae and their application as medicinal foods. Adv. Food Nutr. Res. 2011, 64, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Chiu, L.C.; Ooi, V.E.; Ang, P.O., Jr. A potent antitumor polysaccharide from the edible brown seaweed Hydroclathrus clathratus. Bot. Mar. 2010, 53, 265–274. [Google Scholar] [CrossRef]
- Jiao, L.; Li, X.; Li, T.; Jiang, P.; Zhang, L.; Wu, M.; Zhang, L. Characterization and anti-tumor activity of alkali-extracted polysaccharide from Enteromorpha intestinalis. Int. Immunopharmacol. 2009, 9, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, F.; Zhang, X.W.; Wang, S.C.; Li, M.H.; Lin, L.P.; Ding, J. Grateloupia longifolia polysaccharide inhibits angiogenesis by downregulating tissue factor expression in HMEC-1 endothelial cells. Br. J. Pharmacol. 2006, 148, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Sugahara, T.; Ueno, M.; Fukuta, Y.; Ochi, Y.; Akiyama, K.; Miyazaki, T.; Masuda, S.; Kawakubo, A.; Kato, K. The anti-tumor effect of Euchema serra agglutinin on colon cancer cells in vitro and in vivo. Anticancer Drugs 2006, 17, 943–947. [Google Scholar] [CrossRef]
- Dias, P.F.; Siqueira, J.M., Jr.; Vendruscolo, L.F.; de Jesus Neiva, T.; Gagliardi, A.R.; Maraschin, M.; Ribeiro-do-Valle, R.M. Antiangiogenic and antitumoral properties of a polysaccharide isolated from the seaweed Sargassum stenophyllum. Cancer Chemother. Pharmacol. 2005, 56, 436–446. [Google Scholar] [CrossRef]
- Tang, X.; Li, J.; Xin, X.; Geng, M. A new marine-derived sulfated polysaccharide from brown alga suppresses tumor metastasis both in vitro and in vivo. Cancer Biol. Ther. 2006, 5, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Mishima, T.; Murata, J.; Toyoshima, M.; Fujii, H.; Nakajima, M.; Hayashi, T.; Kato, T.; Saiki, I. Inhibition of tumor invasion and metastasis by calcium spirulan (Ca-SP), a novel sulfated polysaccharide derived from a blue-green alga, Spirulina platensis. Clin. Exp. Metastasis. 1998, 16, 541–550. [Google Scholar] [CrossRef]
- Hsu, H.Y.; Lin, T.Y.; Hwang, P.A.; Tseng, L.M.; Chen, R.H.; Tsao, S.M.; Hsu, J. Fucoidan induces changes in the epithelial to mesenchymal transition and decreases metastasis by enhancing ubiquitin-dependent TGFbeta receptor degradation in breast cancer. Carcinogenesis 2013, 34, 874–884. [Google Scholar] [CrossRef]
- Mizrachi, A.; Shamay, Y.; Shah, J.; Brook, S.; Soong, J.; Rajasekhar, V.K.; Humm, J.L.; Healey, J.H.; Powell, S.N.; Baselga, J.; et al. Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma. Nat. Commun. 2017, 8, 14292. [Google Scholar] [CrossRef]
- Hsu, H.Y.; Lin, T.Y.; Hu, C.H.; Shu, D.T.F.; Lu, M.K. Fucoidan upregulates TLR4/CHOP-mediated caspase-3 and PARP activation to enhance cisplatin-induced cytotoxicity in human lung cancer cells. Cancer Lett. 2018, 432, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ikeguchi, M.; Yamamoto, M.; Arai, Y.; Maeta, Y.; Ashida, K.; Katano, K.; Miki, Y.; Kimura, T. Fucoidan reduces the toxicities of chemotherapy for patients with unresectable advanced or recurrent colorectal cancer. Oncol. Lett. 2011, 2, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.L.; Tai, C.J.; Huang, C.W.; Chang, F.R.; Wang, J.Y. Efficacy of Low-Molecular-Weight Fucoidan as a Supplemental Therapy in Metastatic Colorectal Cancer Patients: A Double-Blind Randomized Controlled Trial. Mar. Drugs 2017, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weelden, G.; Bobinski, M.; Okla, K.; van Weelden, W.J.; Romano, A.; Pijnenborg, J.M.A. Fucoidan Structure and Activity in Relation to Anti-Cancer Mechanisms. Mar. Drugs 2019, 17, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George Kerry, R.P.; Das, G.; Gouda, S.; Kumara Swamy, M.; Kumar Patra, J. Anticancer Potential of Mangrove Plants: Neglected Plant Species of the Marine Ecosystem. In Anticancer Plants: Properties and Applications; Springer Nature: Singapore, 2018; Volume 1, pp. 303–305. [Google Scholar]
- Chakraborty, T.; Bhuniya, D.; Chatterjee, M.; Rahaman, M.; Singha, D.; Chatterjee, B.N.; Datta, S.; Rana, A.; Samanta, K.; Srivastawa, S.; et al. Acanthus ilicifolius plant extract prevents DNA alterations in a transplantable Ehrlich ascites carcinoma-bearing murine model. World J. Gastroenterol. 2007, 13, 6538–6548. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.C.; Yang, S.F.; Liu, S.J.; Kuo, W.H.; Chang, Y.Z.; Hsieh, Y.S. In vitro and in vivo antimetastatic effects of Terminalia catappa L. leaves on lung cancer cells. Food Chem. Toxicol. 2007, 45, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Yang, Y.; Kohler, R.; Giaisi, M.; Witzens-Harig, M.; Liu, D.; Krammer, P.H.; Lin, W.; Li-Weber, M. Mangrove dolabrane-type of diterpenes tagalsins suppresses tumor growth via ROS-mediated apoptosis and ATM/ATR-Chk1/Chk2-regulated cell cycle arrest. Int. J. Cancer 2015, 137, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Jones, W.P.; Lobo-Echeverri, T.; Mi, Q.; Chai, H.; Lee, D.; Soejarto, D.D.; Cordell, G.A.; Pezzuto, J.M.; Swanson, S.M.; Kinghorn, A.D. Antitumour activity of 3-chlorodeoxylapachol, a naphthoquinone from Avicennia germinans collected from an experimental plot in southern Florida. J. Pharm. Pharmacol. 2005, 57, 1101–1108. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Guruvayoorappan, C. Anti-inflammatory and anti-tumor activity of the marine mangrove Rhizophora apiculata. J. Immunotoxicol. 2012, 9, 341–352. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins–Antitumor Polyether Macrolides from a Marine Sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001, 61, 1013–1021. [Google Scholar] [PubMed]
- Lavelle, F.; Zerial, A.; Fizames, C.; Rabault, B.; Curaudeau, A. Antitumor activity and mechanism of action of the marine compound girodazole. Invest. New Drugs 1991, 9, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Motoki, K.; Natori, T.; Uchida, T.; Fukushima, H.; Koezuka, Y. Enhancing effects of agelasphin-11 on natural killer cell activities of normal and tumor-bearing mice. Biol. Pharm. Bull. 1996, 19, 350–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidane, M.; More, M.T.; Pondaven, P.; Jaquot, C.; Riou, D.; Roussakis, C. In vivo effect of pachymatismin, a new marine glycoprotein, on a human non-small-cell lung carcinoma. In Vivo 1997, 11, 185–188. [Google Scholar] [PubMed]
- Copp, B.R.; Fairchild, C.R.; Cornell, L.; Casazza, A.M.; Robinson, S.; Ireland, C.M. Naamidine A is an antagonist of the epidermal growth factor receptor and an in vivo active antitumor agent. J. Med. Chem. 1998, 41, 3909–3911. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, N.; Sato, A.; Hata, T.; Sato, N.; Sasagawa, K.; Kobayashi, T. Cytotoxic scalarane sesterterpenes from a sponge, Hyrtios erecta. J. Nat. Prod. 1998, 61, 468–473. [Google Scholar] [CrossRef]
- Xiong, S.; Pang, H.D.; Fan, J.; Ge, F.; Yang, X.X.; Liu, Q.Y.; Liao, X.J.; Xu, S.H. In vitro and in vivo antineoplastic activity of a novel bromopyrrole and its potential mechanism of action. Br. J. Pharmacol. 2010, 159, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Diez, M.; Guillen-Navarro, M.J.; Pera, B.; Bouchet, B.P.; Martinez-Leal, J.F.; Barasoain, I.; Cuevas, C.; Andreu, J.M.; Garcia-Fernandez, L.F.; Diaz, J.F.; et al. PM060184, a new tubulin binding agent with potent antitumor activity including P-glycoprotein over-expressing tumors. Biochem. Pharmacol. 2014, 88, 291–302. [Google Scholar] [CrossRef]
- Sumii, Y.; Kotoku, N.; Fukuda, A.; Kawachi, T.; Arai, M.; Kobayashi, M. Structure-Activity Relationship and in Vivo Anti-Tumor Evaluations of Dictyoceratin-A and -C, Hypoxia-Selective Growth Inhibitors from Marine Sponge. Mar. Drugs 2015, 13, 7419–7432. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Otte, K.; Alsdorf, W.H.; Hauschild, J.; Lange, T.; Venz, S.; Bauer, C.K.; Bahring, R.; Amann, K.; Mandanchi, R.; et al. Marine compound rhizochalinin shows high in vitro and in vivo efficacy in castration resistant prostate cancer. Oncotarget 2016, 7, 69703–69717. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Liu, G.; Yang, F.; Xing, X.; Li, Y.; Huang, Z.; Yuan, H. Induction of apoptosis and proliferation inhibition of hepatocellular carcinoma by 6-chloro-2-methoxy-N-(phenylmethyl)-9-acridinamine (BA): in vitro and vivo studies. Cancer Cell Int. 2017, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells. Mar. Drugs 2018, 16, 53. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Aurantoside C Targets and Induces Apoptosis in Triple Negative Breast Cancer Cells. Mar. Drugs 2018, 16, 361. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.M.; Rodriguez, A.D.; Berlinck, R.G.; Fusetani, N. Marine pharmacology in 2007-8: Marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous system, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2011, 153, 191–222. [Google Scholar] [CrossRef]
- Urdiales, J.L.; Morata, P.; Nunez De Castro, I.; Sanchez-Jimenez, F. Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from Mediterranean tunicates. Cancer Lett. 1996, 102, 31–37. [Google Scholar] [CrossRef]
- Vervoort, H.; Fenical, W.; Epifanio, R.A. Tamandarins A and B: new cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000, 65, 782–792. [Google Scholar] [CrossRef]
- Edler, M.C.; Fernandez, A.M.; Lassota, P.; Ireland, C.M.; Barrows, L.R. Inhibition of tubulin polymerization by vitilevuamide, a bicyclic marine peptide, at a site distinct from colchicine, the vinca alkaloids, and dolastatin 10. Biochem. Pharmacol. 2002, 63, 707–715. [Google Scholar] [CrossRef]
- Wieczorek, M.; Tcherkezian, J.; Bernier, C.; Prota, A.E.; Chaaban, S.; Rolland, Y.; Godbout, C.; Hancock, M.A.; Arezzo, J.C.; Ocal, O.; et al. The synthetic diazonamide DZ-2384 has distinct effects on microtubule curvature and dynamics without neurotoxicity. Sci. Transl. Med. 2016, 8, 365ra15. [Google Scholar] [CrossRef] [Green Version]
- Sigel, M.M.; Wellham, L.L.; Lichter, W.; Dudeck, L.E.; Gargus, J.L.; Lucas, A.H. Food-Drugs From the Sea: Proceedings. In Frontiers in Natural Product Chemistry; Bentham Science Publishers: Sharjah, UAE, 1969; pp. 281–294. [Google Scholar]
- Rinehart, K.L., Jr.; Gloer, J.B.; Hughes, R.G., Jr.; Renis, H.E.; McGovren, J.P.; Swynenberg, E.B.; Stringfellow, D.A.; Kuentzel, S.L.; Li, L.H. Didemnins: antiviral and antitumor depsipeptides from a caribbean tunicate. Science 1981, 212, 933–935. [Google Scholar] [CrossRef]
- Chittchang, M.; Batsomboon, P.; Ruchirawat, S.; Ploypradith, P. Cytotoxicities and structure-activity relationships of natural and unnatural lamellarins toward cancer cell lines. ChemMedChem 2009, 4, 457–465. [Google Scholar] [CrossRef]
- Schupp, P.; Steube, K.; Meyer, C.; Proksch, P. Anti-proliferative effects of new staurosporine derivatives isolated from a marine ascidian and its predatory flatworm. Cancer Lett. 2001, 174, 165–172. [Google Scholar] [CrossRef]
- Gouiffes, D.; Juge, M.; Grimaud, N.; Welin, L.; Sauviat, M.P.; Barbin, Y.; Laurent, D.; Roussakis, C.; Henichart, J.P.; Verbist, J.F. Bistramide A, a new toxin from the urochordata Lissoclinum bistratum Sluiter: isolation and preliminary characterization. Toxicon 1988, 26, 1129–1136. [Google Scholar] [CrossRef]
- Nazari, M.; Serrill, J.D.; Sikorska, J.; Ye, T.; Ishmael, J.E.; McPhail, K.L. Discovery of Mandelalide E and Determinants of Cytotoxicity for the Mandelalide Series. Org. Lett. 2016, 18, 1374–1377. [Google Scholar] [CrossRef]
- Fontana, A.; Cavaliere, P.; Wahidulla, S.; Naik, C.G.; Cimino, G. A new antitumor isoquinoline alkaloid from the marine nudibranch Jorunna funebris. Tetrahedron 2000, 56, 7305–7308. [Google Scholar] [CrossRef]
- Saito, N.; Tanaka, C.; Koizumi, Y.; Suwanborirux, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A. Chemistry of renieramycins. Part 6: Transformation of renieramycin M into jorumycin and renieramycin J including oxidative degradation products, mimosamycin, renierone, and renierol acetate. Tetrahedron 2004, 60, 3873–3881. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Fujii, Y.; Kizu, H.; Boyd, M.R.; Boettner, F.E.; Doubek, D.L.; Schmidt, J.M.; Chapuis, J.C.; et al. Isolation of Dolastatins 10-15 from the Marine Mollusk Dolabella-Auricularia. Tetrahedron 1993, 49, 9151–9170. [Google Scholar] [CrossRef]
- Sato, M.; Sagawa, M.; Nakazato, T.; Ikeda, Y.; Kizaki, M. A natural peptide, dolastatin 15, induces G2/M cell cycle arrest and apoptosis of human multiple myeloma cells. Int. J. Oncol. 2007, 30, 1453–1459. [Google Scholar] [CrossRef]
- Natural Product Chemistry for Drug Discovery. Rsc. Biomol. Sci. 2010, 1–440.
- Serova, M.; de Gramont, A.; Bieche, I.; Riveiro, M.E.; Galmarini, C.M.; Aracil, M.; Jimeno, J.; Faivre, S.; Raymond, E. Predictive factors of sensitivity to elisidepsin, a novel Kahalalide F-derived marine compound. Mar. Drugs 2013, 11, 944–959. [Google Scholar] [CrossRef] [Green Version]
- Nakao, Y.; Yoshida, W.Y.; Takada, Y.; Kimura, J.; Yang, L.; Mooberry, S.L.; Scheuer, P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004, 67, 1332–1340. [Google Scholar] [CrossRef]
- Aldrich, L.N.; Stoops, S.L.; Crews, B.C.; Marnett, L.J.; Lindsley, C.W. Total synthesis and biological evaluation of tambjamine K and a library of unnatural analogs. Bioorg. Med. Chem. Lett. 2010, 20, 5207–5211. [Google Scholar] [CrossRef]
- Manuel-Manresa, P.; Korrodi-Gregorio, L.; Hernando, E.; Villanueva, A.; Martinez-Garcia, D.; Rodilla, A.M.; Ramos, R.; Fardilha, M.; Moya, J.; Quesada, R.; et al. Novel Indole-based Tambjamine-Analogues Induce Apoptotic Lung Cancer Cell Death through p38 Mitogen-Activated Protein Kinase Activation. Mol. Cancer Ther. 2017, 16, 1224–1235. [Google Scholar] [CrossRef] [Green Version]
- Prendiville, J.; McGown, A.T.; Gescher, A.; Dickson, A.J.; Courage, C.; Pettit, G.R.; Crowther, D.; Fox, B.W. Establishment of a murine leukaemia cell line resistant to the growth-inhibitory effect of bryostatin 1. Br. J. Cancer 1994, 70, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Kraft, A.S.; Woodley, S.; Pettit, G.R.; Gao, F.; Coll, J.C.; Wagner, F. Comparison of the antitumor activity of bryostatins 1, 5, and 8. Cancer Chemother. Pharmacol. 1996, 37, 271–278. [Google Scholar] [CrossRef]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-Lopez, M.; Perez-Sanchez, A.; Galiano, V.; Barrajon-Catalan, E.; Micol, V. An Updated Review on Marine Anticancer Compounds: The Use of Virtual Screening for the Discovery of Small-Molecule Cancer Drugs. Molecules 2017, 22, 37. [Google Scholar] [CrossRef]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total Synthesis of Halichondrin-B and Norhalichondrin-B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Dieras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Lavelle, F.C.; Bayssas, M.; Ahond, A.; Poupat, C.; Pusset, J.; Laurent, D.; Potier, P. Girodazole: "from the lagoon of Noumea to cancer patients". In Proceedings of Third Pacific-Asia Symposium on Biologically Active Natural Products; ORSTOM: Nouméa, New Caledonia, 1992. [Google Scholar]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.H.; Steinmetz, M.O. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. U S A 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.W.; Donia, M.S.; McIntosh, J.A.; Fricke, W.F.; Ravel, J. Origin and variation of tunicate secondary metabolites. J. Nat. Prod. 2012, 75, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, D.W.; Zukoski, C.F. Didemnin B: a new immunosuppressive cyclic peptide with potent activity in vitro and in vivo. Transplantation 1985, 40, 49–56. [Google Scholar] [CrossRef]
- Marco, E.; Martin-Santamaria, S.; Cuevas, C.; Gago, F. Structural basis for the binding of didemnins to human elongation factor eEF1A and rationale for the potent antitumor activity of these marine natural products. J. Med. Chem. 2004, 47, 4439–4452. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.A.; Grubb, D.R.; Lawen, A. Didemnin B induces apoptosis in proliferating but not resting peripheral blood mononuclear cells. Apoptosis 2002, 7, 407–412. [Google Scholar] [CrossRef]
- Dorr, F.A.; Kuhn, J.G.; Phillips, J.; von Hoff, D.D. Phase I clinical and pharmacokinetic investigation of didemnin B, a cyclic depsipeptide. Eur. J. Cancer Clin. Oncol. 1988, 24, 1699–1706. [Google Scholar] [CrossRef]
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullie, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef]
- Cuevas, C.; Francesch, A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009, 26, 322–337. [Google Scholar] [CrossRef]
- Cooper, E.L.; Yao, D. Diving for drugs: tunicate anticancer compounds. Drug. Discov. Today 2012, 17, 636–648. [Google Scholar] [CrossRef]
- Ota, Y.; Chinen, T.; Yoshida, K.; Kudo, S.; Nagumo, Y.; Shiwa, Y.; Yamada, R.; Umihara, H.; Iwasaki, K.; Masumoto, H.; et al. Eudistomin C, an Antitumor and Antiviral Natural Product, Targets 40S Ribosome and Inhibits Protein Translation. Chembiochem 2016, 17, 1616–1620. [Google Scholar] [CrossRef]
- Bailly, C. Anticancer properties of lamellarins. Mar Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef] [Green Version]
- Palanisamy, S.K.; Rajendran, N.M.; Marino, A. Natural Products Diversity of Marine Ascidians (Tunicates; Ascidiacea) and Successful Drugs in Clinical Development. Nat. Prod. Bioprospect. 2017, 7, 1–111. [Google Scholar] [CrossRef] [Green Version]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin is the primary cellular receptor of bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef]
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.T.; Ishmael, J.E.; McPhail, K.L. Mandelalides A-D, cytotoxic macrolides from a new Lissoclinum species of South African tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef] [Green Version]
- Nazari, M.; Serrill, J.D.; Wan, X.; Nguyen, M.H.; Anklin, C.; Gallegos, D.A.; Smith, A.B., 3rd; Ishmael, J.E.; McPhail, K.L. New Mandelalides Expand a Macrolide Series of Mitochondrial Inhibitors. J. Med. Chem. 2017, 60, 7850–7862. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Lefranc, F.; Carbone, M.; Mollo, E.; Gavagnin, M.; Betancourt, T.; Dasari, R.; Kornienko, A.; Kiss, R. Marine Mollusk-Derived Agents with Antiproliferative Activity as Promising Anticancer Agents to Overcome Chemotherapy Resistance. Med. Res. Rev. 2017, 37, 702–801. [Google Scholar] [CrossRef]
- Lane, J.W.; Estevez, A.; Mortara, K.; Callan, O.; Spencer, J.R.; Williams, R.M. Antitumor activity of tetrahydroisoquinoline analogues 3-epi-jorumycin and 3-epi-renieramycin G. Bioorg. Med. Chem. Lett. 2006, 16, 3180–3183. [Google Scholar] [CrossRef] [Green Version]
- Welin, E.R.; Ngamnithiporn, A.; Klatte, M.; Lapointe, G.; Pototschnig, G.M.; McDermott, M.S.J.; Conklin, D.; Gilmore, C.D.; Tadross, P.M.; Haley, C.K.; et al. Concise total syntheses of (-)-jorunnamycin A and (-)-jorumycin enabled by asymmetric catalysis. Science 2019, 363, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Singh, S.B.; Niven, M.L.; Hamel, E.; Schmidt, J.M. Isolation, structure, and synthesis of combretastatins A-1 and B-1, potent new inhibitors of microtubule assembly, derived from Combretum caffrum. J. Nat. Prod. 1987, 50, 119–131. [Google Scholar] [CrossRef]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Kalemkerian, G.P.; Ou, X.L.; Adil, M.R.; Rosati, R.; Khoulani, M.M.; Madan, S.K.; Pettit, G.R. Activity of dolastatin 10 against small-cell lung cancer in vitro and in vivo: induction of apoptosis and bcl-2 modification. Cancer Chemoth. Pharm. 1999, 43, 507–515. [Google Scholar] [CrossRef]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Invest. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef]
- Hamann, M.T.; Scheuer, P.J. Kahalalide-F—A Bioactive Depsipeptide from the Sacoglossan Mollusk Elysia-Rufescens and the Green-Alga Bryopsis Sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Janmaat, M.L.; Rodriguez, J.A.; Jimeno, J.; Kruyt, F.A.; Giaccone, G. Kahalalide F induces necrosis-like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol. Pharmacol. 2005, 68, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Miguel-Lillo, B.; Valenzuela, B.; Peris-Ribera, J.E.; Soto-Matos, A.; Perez-Ruixo, J.J. Population pharmacokinetics of kahalalide F in advanced cancer patients. Cancer Chemother. Pharmacol. 2015, 76, 365–374. [Google Scholar] [CrossRef]
- Jimeno, J.; Lopez-Martin, J.A.; Ruiz-Casado, A.; Izquierdo, M.A.; Scheuer, P.J.; Rinehart, K. Progress in the clinical development of new marine-derived anticancer compounds. Anticancer Drugs 2004, 15, 321–329. [Google Scholar] [CrossRef]
- Waeschenbach, A.; Taylor, P.D.; Littlewood, D.T. A molecular phylogeny of bryozoans. Mol. Phylogenet Evol. 2012, 62, 718–735. [Google Scholar] [CrossRef]
- Carbone, M.; Irace, C.; Costagliola, F.; Castelluccio, F.; Villani, G.; Calado, G.; Padula, V.; Cimino, G.; Lucas Cervera, J.; Santamaria, R.; et al. A new cytotoxic tambjamine alkaloid from the Azorean nudibranch Tambja ceutae. Bioorg. Med. Chem. Lett. 2010, 20, 2668–2670. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Zonder, J.A.; Shields, A.F.; Zalupski, M.; Chaplen, R.; Heilbrun, L.K.; Arlauskas, P.; Philip, P.A. A phase II trial of bryostatin 1 in the treatment of metastatic colorectal cancer. Clin. Cancer Res. 2001, 7, 38–42. [Google Scholar]
- Morgan, R.J., Jr.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: a California cancer consortium study. Invest. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef] [Green Version]
- van der Hem, K.G.; Drager, A.M.; Huijgens, P.C.; Tol, C.; Deville, W.; Langenhuijsen, M.M. The differentiation inducing effect of bryostatin 5 on human myeloid blast cells is potentiated by vitamin D3. Leukemia 1994, 8, 266–273. [Google Scholar]
- Evans, J.S.; Musser, E.A.; Mengel, G.D.; Forsblad, K.R.; Hunter, J.H. Antitumor activity of 1-beta-D-arainofuranosylcytosine hydrochloride. Proc. Soc. Exp. Biol. Med. 1961, 106, 350–353. [Google Scholar] [CrossRef]
- El-Subbagh, H.I.; Al-Badr, A.A. Chapter 2 cytarabine. Profiles Drug Subst. Excip. Relat. Methodol. 2009, 34, 37–113. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Lisyanskaya, A.S.; Makhson, A.N.; Rolski, J.; et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J. Clin. Oncol. 2010, 28, 3107–3114. [Google Scholar] [CrossRef]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother. Pharmacol. 2016, 77, 663–671. [Google Scholar] [CrossRef]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1342. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef]
- Schoffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Poncet, J. The dolastatins, a family of promising antineoplastic agents. Curr. Pharm. Des. 1999, 5, 139–162. [Google Scholar]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fernandez, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernandez-Sousa, J.M.; Munoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef] [Green Version]
- Losada, A.; Munoz-Alonso, M.J.; Garcia, C.; Sanchez-Murcia, P.A.; Martinez-Leal, J.F.; Dominguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef]
- Depenbrock, H.; Peter, R.; Faircloth, G.T.; Manzanares, I.; Jimeno, J.; Hanauske, A.R. In vitro activity of aplidine, a new marine-derived anti-cancer compound, on freshly explanted clonogenic human tumour cells and haematopoietic precursor cells. Br. J. Cancer 1998, 78, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Caers, J.; Menu, E.; De Raeve, H.; Lepage, D.; Van Valckenborgh, E.; Van Camp, B.; Alvarez, E.; Vanderkerken, K. Antitumour and antiangiogenic effects of Aplidin in the 5TMM syngeneic models of multiple myeloma. Br. J. Cancer 2008, 98, 1966–1974. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Alvarez, S.; Pardal, E.; Sanchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martin, A. Plitidepsin: design, development, and potential place in therapy. Drug Des Devel Ther 2017, 11, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Huang, S.Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Martinez, S.; Extremera, S.; et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann. Hematol. 2019, 98, 2139–2150. [Google Scholar] [CrossRef] [Green Version]
- Janssens, A.; Boogaerts, M.; Verhoef, G. Development of fludarabine formulations in the treatment of chronic lymphocytic leukemia. Drug Des. Devel. Ther. 2009, 3, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.H.; Johnson, J.R.; Justice, R.; Pazdur, R. FDA drug approval summary: nelarabine (Arranon) for the treatment of T-cell lymphoblastic leukemia/lymphoma. Oncologist 2008, 13, 709–714. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Luis, G.P.-A.; Jose Manuel Trigo, P.; Benjamin, B.; Victor, M.; Rafael, L.; Maria Angeles, S.; Santiago Ponce, A.; Cristian Marcelo, F.; Mariano, S.; Carmen Maria, K.; et al. Efficacy and safety profile of lurbinectedin in second-line SCLC patients: Results from a phase II single-agent trial. J. Clin. Oncol. 2019, 37, 8506. [Google Scholar] [CrossRef]
- Sorolla, A.; Yeramian, A.; Dolcet, X.; Perez de Santos, A.M.; Llobet, D.; Schoenenberger, J.A.; Casanova, J.M.; Soria, X.; Egido, R.; Llombart, A.; et al. Effect of proteasome inhibitors on proliferation and apoptosis of human cutaneous melanoma-derived cell lines. Br. J. Dermatol. 2008, 158, 496–504. [Google Scholar] [CrossRef]
- Sorolla, A.; Yeramian, A.; Valls, J.; Dolcet, X.; Bergada, L.; Llombart-Cussac, A.; Marti, R.M.; Matias-Guiu, X. Blockade of NFkappaB activity by Sunitinib increases cell death in Bortezomib-treated endometrial carcinoma cells. Mol. Oncol. 2012, 6, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Yeramian, A.; Sorolla, A.; Velasco, A.; Santacana, M.; Dolcet, X.; Valls, J.; Abal, L.; Moreno, S.; Egido, R.; Casanova, J.M.; et al. Inhibition of activated receptor tyrosine kinases by Sunitinib induces growth arrest and sensitizes melanoma cells to Bortezomib by blocking Akt pathway. Int J Cancer 2012, 130, 967–978. [Google Scholar] [CrossRef]
- Marti, R.M.; Sorolla, A.; Yeramian, A. New therapeutic targets in melanoma. Actas Dermosifiliogr. 2012, 103, 579–590. [Google Scholar] [CrossRef]
- Ortega, E.; Marti, R.M.; Yeramian, A.; Sorolla, A.; Dolcet, X.; Llobet, D.; Abal, L.; Santacana, M.; Pallares, J.; Llombart-Cussac, A.; et al. Targeted therapies in gynecologic cancers and melanoma. Semin. Diagn. Pathol. 2008, 25, 262–273. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Encinas, M.; Pallares, J.; Cabero, A.; Schoenenberger, J.A.; Comella, J.X.; Matias-Guiu, X. Proteasome inhibitors induce death but activate NF-kappaB on endometrial carcinoma cell lines and primary culture explants. J. Biol. Chem. 2006, 281, 22118–22130. [Google Scholar] [CrossRef] [Green Version]
- Llobet, D.; Eritja, N.; Encinas, M.; Sorolla, A.; Yeramian, A.; Schoenenberger, J.A.; Llombart-Cussac, A.; Marti, R.M.; Matias-Guiu, X.; Dolcet, X. Antioxidants block proteasome inhibitor function in endometrial carcinoma cells. Anticancer Drugs 2008, 19, 115–124. [Google Scholar] [CrossRef]
- Belarbi el, H.; Contreras Gomez, A.; Chisti, Y.; Garcia Camacho, F.; Molina Grima, E. Producing drugs from marine sponges. Biotechnol. Adv. 2003, 21, 585–598. [Google Scholar] [CrossRef]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, J.R.; McFall, G.; Zea, S. Does the odor from sponges of the genus Ircinia protect them from fish predators? J. Chem. Ecol. 2002, 28, 1103–1115. [Google Scholar] [CrossRef]
- Piel, J.; Hui, D.; Wen, G.; Butzke, D.; Platzer, M.; Fusetani, N.; Matsunaga, S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc. Natl. Acad. Sci. USA 2004, 101, 16222–16227. [Google Scholar] [CrossRef] [Green Version]
- Sears, M.A.; Gerhart, D.J.; Rittschof, D. Antifouling agents from marine spongeLissodendoryx isodictyalis carter. J. Chem. Ecol. 1990, 16, 791–799. [Google Scholar] [CrossRef]
- Tsoukatou, M.; Marechal, J.P.; Hellio, C.; Novakovic, I.; Tufegdzic, S.; Sladic, D.; Gasic, M.J.; Clare, A.S.; Vagias, C.; Roussis, V. Evaluation of the activity of the sponge metabolites avarol and avarone and their synthetic derivatives against fouling micro- and macroorganisms. Molecules 2007, 12, 1022–1034. [Google Scholar] [CrossRef] [Green Version]
- Webster, N.S.; Taylor, M.W. Marine sponges and their microbial symbionts: love and other relationships. Environ. Microbiol. 2012, 14, 335–346. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural Products as a Foundation for Drug Discovery. Curr. Protoc. Pharmacol. 2009, 46, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Radjasa, O.K.; Vaske, Y.M.; Navarro, G.; Vervoort, H.C.; Tenney, K.; Linington, R.G.; Crews, P. Highlights of marine invertebrate-derived biosynthetic products: their biomedical potential and possible production by microbial associants. Bioorg. Med. Chem. 2011, 19, 6658–6674. [Google Scholar] [CrossRef] [Green Version]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Nandy, J.P.; Prakesch, M.; Khadem, S.; Reddy, P.T.; Sharma, U.; Arya, P. Advances in solution- and solid-phase synthesis toward the generation of natural product-like libraries. Chem. Rev. 2009, 109, 1999–2060. [Google Scholar] [CrossRef]
- Kumar, K.; Waldmann, H. Synthesis of natural product inspired compound collections. Angew. Chem. Int. Ed. Engl. 2009, 48, 3224–3242. [Google Scholar] [CrossRef]
- Uemura, D.; Takahashi, K.; Yamamoto, T.; Katayama, C.; Tanaka, J.; Okumura, Y.; Hirata, Y. Norhalichondrin-a–an Antitumor Polyether Macrolide from a Marine Sponge. J. Am. Chem. Soc. 1985, 107, 4796–4798. [Google Scholar] [CrossRef]
- Ledford, H. Complex synthesis yields breast-cancer therapy. Nature 2010, 468, 608–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huyck, T.K.; Gradishar, W.; Manuguid, F.; Kirkpatrick, P. Eribulin mesylate. Nat. Rev. Drug Discov. 2011, 10, 173–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbach, M.A.; Clardy, J. One pathway, many products. Nat. Chem. Biol. 2007, 3, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Lam, K.S. Generating a generation of proteasome inhibitors: from microbial fermentation to total synthesis of salinosporamide a (marizomib) and other salinosporamides. Mar. Drugs 2010, 8, 835–880. [Google Scholar] [CrossRef] [Green Version]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol 2012, 19, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Sosa-Hernandez, J.E.; Escobedo-Avellaneda, Z.; Iqbal, H.M.N.; Welti-Chanes, J. State-of-the-Art Extraction Methodologies for Bioactive Compounds from Algal Biome to Meet Bio-Economy Challenges and Opportunities. Molecules 2018, 23, 2953. [Google Scholar] [CrossRef] [Green Version]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.C.; Madeira, C.; Brandao, C.A.; Puga, J.; Calado, R. Bioprospecting of marine invertebrates for new natural products–a chemical and zoogeographical perspective. Molecules 2012, 17, 9842–9854. [Google Scholar] [CrossRef]
- Danovaro, R.; Carugati, L.; Berzano, M.; Cahill, A.E.; Carvalho, S.; Chenuil, A.; Corinaldesi, C.; Cristina, S.; David, R.; Dell’Anno, A.; et al. Implementing and Innovating Marine Monitoring Approaches for Assessing Marine Environmental Status. Front. Mar. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Cragg, G.M.; Katz, F.; Newman, D.J.; Rosenthal, J. The impact of the United Nations Convention on Biological Diversity on natural products research. Nat. Prod. Rep. 2012, 29, 1407–1423. [Google Scholar] [CrossRef]
- Maier, M.E. Structural revisions of natural products by total synthesis. Nat. Prod. Rep. 2009, 26, 1105–1124. [Google Scholar] [CrossRef] [PubMed]
- Iyer, U.; Kadambi, V.J. Antibody drug conjugates–Trojan horses in the war on cancer. J. Pharm. Toxicol Methods 2011, 64, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, W.F.; Enriquez, R.G. Choosing the best pulse sequences, acquisition parameters, postacquisition processing strategies, and probes for natural product structure elucidation by NMR spectroscopy. J. Nat. Prod. 2002, 65, 221–244. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Fenner, A.M. Drug discovery from marine microbes. Microb. Ecol. 2013, 65, 800–806. [Google Scholar] [CrossRef]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef]
- Piel, J. Metabolites from symbiotic bacteria. Nat. Prod. Rep. 2009, 26, 338–362. [Google Scholar] [CrossRef]
- Penesyan, A.; Kjelleberg, S.; Egan, S. Development of novel drugs from marine surface associated microorganisms. Mar. Drugs 2010, 8, 438. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.; Martin, G.D.A. Identification and bioactive potential of marine microorganisms from selected Florida coastal areas. Microbiologyopen 2017, 6. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Muller, E.E.; Hu, Y.O.; Lebrun, L.A.; Roume, H.; Lundin, D.; Wilmes, P.; Andersson, A.F. Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PLoS ONE 2014, 9, e95567. [Google Scholar] [CrossRef]
- Bucklin, A.; Lindeque, P.K.; Rodriguez-Ezpeleta, N.; Albaina, A.; Lehtiniemi, M. Metabarcoding of marine zooplankton: prospects, progress and pitfalls. J. Plankton Res. 2016, 38, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Cole, J.R.; Zhang, Q.; Brown, C.T.; Tiedje, J.M. Microbial Community Analysis with Ribosomal Gene Fragments from Shotgun Metagenomes. Appl. Environ. Microbiol. 2016, 82, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Sorolla, A.; Ho, D.; Wang, E.; Evans, C.W.; Ormonde, C.F.; Rashwan, R.; Singh, R.; Iyer, K.S.; Blancafort, P. Sensitizing basal-like breast cancer to chemotherapy using nanoparticles conjugated with interference peptide. Nanoscale 2016, 8, 9343–9353. [Google Scholar] [CrossRef]
- Clemons, T.D.; Singh, R.; Sorolla, A.; Chaudhari, N.; Hubbard, A.; Iyer, K.S. Distinction between Active and Passive Targeting of Nanoparticles Dictate Their Overall Therapeutic Efficacy. Langmuir 2018, 34, 15343–15349. [Google Scholar] [CrossRef] [Green Version]
- Sorolla, A.; Wang, E.; Clemons, T.D.; Evans, C.W.; Plani-Lam, J.H.; Golden, E.; Dessauvagie, B.; Redfern, A.D.; Swaminathan-Iyer, K.; Blancafort, P. Triple-hit therapeutic approach for triple negative breast cancers using docetaxel nanoparticles, EN1-iPeps and RGD peptides. Nanomedicine 2019, 20, 102003. [Google Scholar] [CrossRef]
- Sorolla, A.; Wang, E.; Golden, E.; Duffy, C.; Henriques, S.T.; Redfern, A.D.; Blancafort, P. Precision medicine by designer interference peptides: applications in oncology and molecular therapeutics. Oncogene 2019. [Google Scholar] [CrossRef] [Green Version]
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk. Res. 2009, 33, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J Clin Oncol 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Beltran, A.S.; Graves, L.M.; Blancafort, P. Novel role of Engrailed 1 as a prosurvival transcription factor in basal-like breast cancer and engineering of interference peptides block its oncogenic function. Oncogene 2014, 33, 4767–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Sorolla, A.; Cunningham, P.T.; Bogdawa, H.M.; Beck, S.; Golden, E.; Dewhurst, R.E.; Florez, L.; Cruickshank, M.N.; Hoffmann, K.; et al. Tumor penetrating peptides inhibiting MYC as a potent targeted therapeutic strategy for triple-negative breast cancers. Oncogene 2019, 38, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Ponnappan, N.; Budagavi, D.P.; Yadav, B.K.; Chugh, A. Membrane-active peptides from marine organisms--antimicrobials, cell-penetrating peptides and peptide toxins: applications and prospects. Probiotics Antimicrob. Proteins 2015, 7, 75–89. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound Name | Marine Organism | Species Name | Active Derivative | Cancer Model | In Vitro | In Vivo | IC50 In Vitro | Route of Administration In Vivo | Dose Used In Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Pyrroloformamide | Actinobacteria | Streptomyces sp. | Pyrrolinonodithiol | Human prostate cancer cell line PC-3M | ✓ | ✗ | 1.67 nM | N/A | N/A | [20] |
| Cromomycin A2 | Actinobacteria | Streptomyces sp. | Aureolic acid | Human melanoma cell line MALME-3M | ✓ | ✗ | 16.7 nM | N/A | N/A | [21] |
| Anthracyclinone 4 | Bacteria | Micromonospora sp. | Anthracyclinone | Human colon adenocarcinoma cell line HCT-8 | ✓ | ✗ | 6.2 µM | N/A | N/A | [22] |
| Coibamide-A | Cyanobacteria | Leptolyngbya sp. | Cyclic depsipeptide | Human lung cancer cell line NCI-H460 and mouse neuro-2a cells | ✓ | ✗ | < 23 nM | N/A | N/A | [23] |
| Lucentamycins A | Bacteria | Nocardiopsis lucentensis | Non-ribosomal peptides | Human colon carcinoma cell line HCT-116 | ✓ | ✗ | 0.20 µM | N/A | N/A | [25] |
| Mixirins A, B & C | Bacteria | Bacillus sp. | Non-ribosomal peptides | Human colon carcinoma cell line HCT-116 | ✓ | ✗ | A: 0.68, B: 1.6, and C: 1.3 mg/mL | N/A | N/A | [26] |
| Ohmyungsamycins A&B | Actinobacteria | Streptomyces sp. | Non-ribosomal peptides | Human colon carcinoma cell line HCT-116 | ✓ | ✗ | A: 0.359 µM, B: 12.4 µM | N/A | N/A | [27] |
| Human lung cancer cell line A549 | ✓ | ✗ | A: 0.551 µM, B: 15.6 µM | N/A | N/A | |||||
| Human stomach cancer cell line SNU-638 | ✓ | ✗ | A: 0.532 µM, B: 13.5 µM | N/A | N/A | |||||
| Human triple negative breast cancer cell line MDA-MB-231 | ✓ | ✗ | A: 0.688 µM, B: 12.7 µM | N/A | N/A | |||||
| Human hepatic adenocarcinoma cancer cell line SK-HEP-1 | ✓ | ✗ | A: 0.816 µM, B: 16.8 µM | N/A | N/A | |||||
| Urukthapelstatin A | Actinobacteria | Mechercharimyces asporophorigenens | Non ribosomal peptides | Human lung cancer lines A549, DMS114, and NCIH460 | ✓ | ✗ | A519: 12 nM | N/A | N/A | [28] |
| Human ovarian cancer cell lines OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8, and SK-OV3 | ✓ | ✗ | 0.828–0.846 nM for the rest | N/A | N/A | |||||
| Human breast cancer cell line MCF-7 | ✓ | ✗ | N/A | N/A | ||||||
| Symplostatin 1 | Cyanobacteria | Symploca hydnoides | Linear pentapeptide | Human colon adenocarcinoma cell line LoVo | ✓ | ✗ | 0.34-0.50 nM | N/A | N/A | [16] |
| HeLa-derived cell line KB | ✓ | ✗ | 0.15-0.20 nM | N/A | N/A | |||||
| Early stage colon adenocarcinoma #38 | ✗ | ✓ | N/A | i.v. | 3 mg/Kg | |||||
| Early stage mammary adenocarcinoma 16/C | ✗ | ✓ | N/A | i.v. | 1.25 mg/Kg | |||||
| Human breast cancer cell line MDA-MB-435 | ✓ | ✗ | 0.15 nM | N/A | N/A | [17] | ||||
| Human ovarian cancer cell line SK-OV-3 | ✓ | ✗ | 0.09 nM | N/A | N/A | |||||
| Multidrug resistant human ovarian cancer cell line NCI/ADR | ✓ | ✗ | 2.9 nM | N/A | N/A | |||||
| Early stage colon adenocarcinoma #38 | ✗ | ✓ | N/A | i.v. | 3 mg/Kg | |||||
| Early stage mammary adenocarcinoma 16/C | ✗ | ✓ | N/A | i.v. | 0.5, 0.25 mg/Kg | |||||
| TZT-1027 | Cyanobacteria | Symploca sp. | Synthetic tetrapeptide. Dolastatin 10 derivative | Murine leukemia P338, melanoma B16, colon cancer colon 26 and sarcoma M5076 allografts | ✗ | ✓ | N/A | i.p. and i.v. | 0.125–3 mg/Kg | [18] |
| Human lung cancer LX-1 and breast carcinoma MX-1 xenografts | ✗ | ✓ | N/A | i.v. | 0.5–2 mg/Kg |
| Compound Name | Marine Organism | Species Name | Active Derivative | Cancer Model | In Vitro | In Vivo | IC50 in Vitro | Route of Administration In Vivo | Dose Used In Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Brocazine G | Fungus | Penicilliu m brocae MA-231 | Disulfide- bridged diketopip erazines | Human ovarian cancer cell lines A2780 and A2780-cisplatin resistant | ✓ | ✗ | A2780: 664 nM A2780 (CisR): 664 nM | N/A | N/A | [32] |
| Compound 2 | Fungus | Penicilliu m sp. FJ-1 | Sesquiterp enoid | Human osteosarcoma cell line MG- 63 and MG-63 xenografts | ✓ | ✓ | 55 nM | i.g. | 10 and 30 mg/Kg | [33] |
| Astaxanthin | Microalgae | Haematoc occus pluvialis | Keto- carotenoid | Induced colonic pre- neoplastic progression in rats induced by 1,2 dimethylhydrazine | ✗ | ✓ | N/A | Orally | 15 mg/Kg | [39] |
| Chlorella sorokiniana extracts | Microalgae | Chlorella sorokinia na | Not specified | Human lung adenocarcinoma cell lines A549 and CL1-5 xenograft | ✓ | ✓ | A549: >40% cell death: 50 ng/mL CL1-5: >70% cell death: 250 ng/mL | Orally | 50 mg/Kg | [40] |
| H3-a1 | Macroalgae | Hydroclat hrus clathratus | Sulfated polysacch aride | Human acute promyelocytic leukemia cell line HL-60, human breast carcinoma cell line MCF-7. and human hepatocarcinoma cancer cell lines | ✓ | ✓ | Not foun d | i.p. | 20 and 50 mg/kg | [41] |
| Murine sarcoma S180 allograft | ✓ | ✓ | Not found | i.p. | 20 and 50 mg/kg | |||||
| DAEB | Macroalgae | Enteromorpha intestinalis | Sulfated polysaccharide | Murine sarcoma S180 cells and S180 allograft | ✓ | ✓ | 5.6% cell death: 800 µg/mL | i.g. | 100, 200, and 400 mg/kg | [43] |
| Grateloupia longifolia poly- saccharide (GLP) | Macroalgae | Grateloupia longifolia | Sulfated polysacchar ide | Human microvascular endothelial cell line | ✓ | ✗ | 0.86 mg/mL | N/A | N/A | [44] |
| HMEC-1 | ||||||||||
| Human umbilical vein endothelial cell line HUVEC | ✓ | ✗ | 0.64 mg/mL | N/A | N/A | |||||
| Murine fibroblast cell line NIH-3T3 | ✓ | ✗ | 1.01 mg/mL | N/A | N/A | |||||
| Human breast cancer cell line MDA-MB-435 | ✓ | ✗ | 1.77 mg/mL | N/A | N/A | |||||
| Human gastric cancer cell line MKN-28 | ✓ | ✗ | 1.66 mg/mL | N/A | N/A | |||||
| Human colon cancer cell line HCT-116 | ✓ | ✗ | 1.42 mg/mL | N/A | N/A | |||||
| Human ovarian cancer cell line SK-OV-3 | ✓ | ✗ | 2.65 mg/mL | N/A | N/A | |||||
| Murine sarcoma cell line S180 and S180 allograft | ✓ | ✓ | 1.72 mg/mL | i.v. | 200 mg/Kg | |||||
| Eucheuma serra aggluttinin | Macroalgae | Eucheuma serra | Sulfated polysaccharide | Murine colon cancer cell line Colon26 and Colon26 allograft | ✓ | ✓ | >10 µg/mL | i.v. | 400 µg | [45] |
| SargA | Macroalgae | Sargassum stenophyllum | Sulfated polysaccharide | Murine melanoma cell line B16F10 and B16F10 allograft | ✓ | ✓ | <200 µg/well | s.c. | 1.5 or 150 µg | [46] |
| Marine-derived sulfated poly- saccharide (MSP) | Macroalgae | Not specified | Sulfated polysaccharide | Human breast carcinoma cell line MDA-MB-231 and murine Lewis lung carcinoma | ✓ | ✓ | >200 µg/mL | i.p. | 10 –80 mg/Kg | [47] |
| Ca-SP | Macroalgae | Spirulina platensis | Sulfated polysaccharide | Murine melanoma lung metastasis model B16- BL6 | ✗ | ✓ | N/A | i.v. | 100 µg | [48] |
| Fucoidan | Macroalgae | Not specified | Sulfated polysaccharide | Human breast carcinoma cell lines 4T1 and MDA- MB-231, and 4T1 xenograft | ✓ | ✓ | >120 µ/mL | Not specified | 0.25 mg | [51] |
| Human head and neck carcinoma cell line Cal- 33, Cal-33 xenograft and PDX H22 | ✗ | ✓ | N/A | i.v. | 7, 25, and 50 mg/Kg | [50] | ||||
| Murine Lewis lung carcinoma cell line LLC1 and LLC1 allograft | ✓ | ✓ | <6.25 µg/mL | Orally | 15 mg/kg | [51] |
| Compound Name | Marine Organism | Species Name | Active Derivative | Cancer Model | In Vitro | In Vivo | IC50 in Vitro | Route of Administration in Vivo | Dose Used in Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| ALE | Higher plant | Acanthus ilicifolius Linn. | Not specified | Murine Ehrlich ascites carcinoma | ✗ | ✓ | N/A | i.p. | 2.5 mg/Kg | [56] |
| TCE | Higher plant | Terminalia catappa L. | Not specified | Human lung adenocarcinoma cell line A549 | ✓ | ✗ | >100 µg/mL | N/A | N/A | [57] |
| Murine Lewis lung carcinoma cell line LLC and LLC allograft | ✓ | ✓ | 14.5 µg/mL | Orally | 3 g/Kg | |||||
| Tagalsin C | Higher plant | Ceriops tagal | Dolabrane-type of diterpene | Human T cell leukemia cell lines Jurkat, SupT1, and Molt-4 | ✓ | ✗ | <2.5 µM | N/A | N/A | [58] |
| Human myeloma cell lines U-266 and PRMI-8266 | ✓ | ✗ | <2.5 µM | N/A | N/A | |||||
| Human lymphoma cell lines L1236 and KM-H2 | ✓ | ✗ | <2.5 µM | N/A | N/A | |||||
| T cells from acute myeloid leukemia patients | ✓ | ✗ | >0.5 µM | N/A | N/A | |||||
| Human T cell leukemia line CEM and CEM xenograft | ✓ | ✓ | < 0.5 µM | i.p. | 50 mg/Kg | |||||
| 3-chlorodeoxylapachol | Higher plant | Avicennia germinans | Naphthoquinone | Human colon cancer cell line Col2 | ✓ | ✗ | 3.7 µg/mL | N/A | N/A | [59] |
| Human prostate cancer cell line LNCaP | ✓ | ✗ | 4.1 µg/mL | N/A | N/A | |||||
| Human lung cancer cell line Lu1 | ✓ | ✗ | 8.3 µg/mL | N/A | N/A | |||||
| Human telomerase reverse transcriptase- retinal pigment epithelium hTERT-RPE1 | ✓ | ✗ | 5 µg/mL | N/A | N/A | |||||
| Human oralepidermoid carcinoma cell line KB and KB xenograft | ✓ | ✓ | 3.2 µg/mL | i.p. | 5 mg/Kg | |||||
| R. apiculata extract | Higher plant | Rhyzophora apiculata | Not specified | Murine melanoma mice model B16F10 | ✗ | ✓ | N/A | i.p. | 10 mg/Kg | [60] |
| Compound Name | Marine Organis | Species Name | Active Derivative | Cancer Model | In Vitro | In Vivo | IC50 in Vitro | Route of Administration In Vivo | Dose used In Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Halichondrin B | Sponge | Halichondria okadai | Polyether macrolide | Murine melanoma cell line B16 and B16 allograft | ✓ | ✓ | 0.093 ng/m L | i.p. and i.v. | 2.5–20 µg/Kg | [61] |
| Murine leukemia cell lines P338 and L-1210 and P338 and L-1210 allografts | ✗ | ✓ | N/A | i.p. | 1.25–100 µg/Kg | |||||
| ER-076349 and ER-086526 | Sponge | synthetically synthesized from Halichondrin B | Macrocyc lic ketone | Human promyelocytic leukemia cell line HL-60 | ✓ | ✗ | 0.41 nM | N/A | N/A | [62] |
| Human histiocytic lymphoma cell line U937 | ✓ | ✗ | 0.22 nM | N/A | N/A | |||||
| Human prostate cancer cell line LNCaP | ✓ | ✗ | 0.25 nM | N/A | N/A | |||||
| ER-076349 and ER-086526 | Sponge | synthetically synthesized from Halichondrin B | Macrocyclic ketone | Human prostate cancer cell line DU 145 | ✓ | ✗ | 0.70 nM | N/A | N/A | [62] |
| Human colon cancer cell line DLD-1 | ✓ | ✗ | 0.75 nM | N/A | N/A | |||||
| Human breast cancer cell line MDA-MB-435 and xenograft | ✓ | ✓ | 0.14 nM | i.v. | 0.25–1 mg/Kg | |||||
| Human colon cancer cell line COLO205 and COLO205 xenograft | ✓ | ✓ | 0.41 nM | i.p. | 0.125–0.5 mg/Kg | |||||
| Human melanoma cell line LOX and LOX xenograft | ✓ | ✓ | 0.76 nM | i.p. | 0.1–0.5 mg/Kg | |||||
| Human ovarian cancer cell line NIH:OVCAR-3 and NIH:OVCAR-3 xenograft | ✗ | ✓ | N/A | i.v. | 0.125–1 mg/Kg | |||||
| Girodazole | Sponge | Pseudaxinyssa cantharella | (1S,2S)-3-amino-1-(2-amino-1H-imidazol-5- yl)-2-chloropropan-1- ol;dihydrochloride | Murine leukemia cell line P388 and P338 and L1210 allografts | ✓ | ✓ | Not found | i.p. | Not found | [63] |
| Murine mammary adenocarcinoma 16/C allograft | ✗ | ✓ | N/A | s.c. | Not found | |||||
| Murine M5076 histiocytosarcoma | ✗ | ✓ | N/A | i.v. | Not found | |||||
| Agelasphin-11 | Sponge | Agelas mauritianus | Galactosylceramide | Murine melanoma model B16 | ✗ | ✓ | N/A | i.v. | 0.1 mg/Kg | [64] |
| Pachymatismin | Sponge | Pachymatisma johnstonii | Glycoprotein | Human non-small cell lung cancer cell line N6 and N6 xenograft | ✓ | ✓ | Not found | Not found | Not mentioned | [65] |
| Naamidine | Sponge | Fijian Leucetta | 2-aminoimidazole alkaloid | Human squamous cell carcinoma A431 xenograft | ✗ | ✓ | N/A | Not found | 25 and 50 mg/Kg | [66] |
| Scalarane sesterterpne 1 | Sponge | Hyrtios erecta | Sesterterpene | Murine lymphatic leukaemia cell line P338 and P338 allograft | ✓ | ✓ | 14.5 ng/mL | i.p. | 0.5–8 mg/Kg | [67] |
| Human gastric cancer cell line MKN-1 | ✓ | ✗ | 57.7 ng/mL | N/A | N/A | |||||
| Human gastric cancer cell lines MKN-7 and MKN-74 | ✓ | ✗ | 56 and 36.8 ng/mL | N/A | N/A | |||||
| B6. Derivative of Aldisin | Sponge | Polymistia sp. | Bromopyrrole | Human nasopharyngeal carcinoma cell line CNE | ✓ | ✗ | 17.18 µg/mL | N/A | N/A | [68] |
| Human breast carcinoma cell line MCF-7 | ✓ | ✗ | 11.30 µg/mL | N/A | N/A | |||||
| Human hepatic carcinoma cell line HepG2 | ✓ | ✗ | 15.30 µg/mL | N/A | N/A | |||||
| Human colon carcinoma cell line Lovo | ✓ | ✗ | 3.83 µg/mL | N/A | N/A | |||||
| Human hepatocarcinoma cell line BEL-7402 | ✓ | ✗ | 10.98 µg/mL | N/A | N/A | |||||
| Human cervical epithelial carcinoma cell line HeLa | ✓ | ✗ | 5.46 µg/mL | N/A | N/A | |||||
| Murine sarcoma S180 and hepatocarcinoma H22 allografts | ✗ | ✓ | N/A | i.g. | 40, 60, and 80 mg/Kg | |||||
| Plocabulin or PM060184 | Sponge | Lithoplocamia lithistoides | Polyketide | Human ovarian cancer cell lines IGROV-1 and IGROV-1/ET | ✓ | ✗ | 0.4 and 4 nM | N/A | N/A | [69] |
| Human ovarian cancer cell lines A2780 and A2780/Dox | ✓ | ✗ | 2.5 and 17 nM | N/A | N/A | |||||
| Human colon carcinoma cell lines Lovo and Lovo/Dox | ✓ | ✗ | 0.5 and 5 nM | N/A | N/A | |||||
| Human breast carcinoma MDA-MB-231 xenograft | ✗ | ✓ | N/A | i.v. | 16 mg/Kg | |||||
| Human colon carcinoma HCT-116 xenograft | ✗ | ✓ | N/A | i.v. | 16 mg/Kg | |||||
| Human gastric cancer NGC-27 xenograft | ✗ | ✓ | N/A | i.v. | 16 mg/Kg | |||||
| Human non-small cell lung cancer H-460 xenograft | ✗ | ✓ | N/A | i.v. | 16 mg/Kg | |||||
| Human prostate cancer 22RV1 xenograft | ✗ | ✓ | N/A | i.v. | 16 mg/Kg | |||||
| Human renal cancer Caki-1 xenograft | ✗ | ✓ | N/A | i.v. | 16 mg/Kg | |||||
| Dictyoceratin-A and -C | Sponge | Dactylospongia elegans | Sesquiterpene phenol | Murine sarcoma cell line S180 | ✗ | ✓ | N/A | Orally | 10–50 mg/Kg | [70] |
| Rhizochalinin | Sponge | Rhizochalina incrustata | Sphingolipid-like | Human prostate cancer cell lines DU145, LNCaP, and VCaP | ✓ | ✗ | DU145, LNCaP: <1.5µM, VCaP: <0.5 µM | N/A | N/A | [71] |
| Human prostate cancer cell PC-3 and PC-3 and 22Rv1 xenografts | ✓ | ✓ | <1.5 µM | i.p. | 1.8 mg/Kg | |||||
| BA | Sponge | Not specified | Acridinamine | Human liver carcinoma cell line SMMC-7221 and SMMC-7221 xenograft | ✓ | ✓ | <16 µM | i.p. | 5 and 10 mg/Kg | [72] |
| Crambescidin 800 | Sponge | Monachora viridis | Not specified | Human breast carcinoma cell line SUM149PT, SUM159PT, MDA-MB- 231, MCF-7, and ZR-75-1 | ✓ | ✗ | 6.02, 3.42, 5, 4.72, 8.09 µM, respectively | N/A | N/A | [73] |
| Aurantoside C | Sponge | Manihinea lynbeazleyae | Not specified | Human breast carcinoma cell line SUM149PT, SUM159PT, MDA-MB- 231, MCF-7, ZR-75-1, and T47D | ✓ | ✗ | 0.81, 0.56, 0.61, 1.15, 1.91, 2.45 µM, respectively | N/A | N/A | [74] |
| Compound Name | Marine Organism | Species Name | Active Derivative | Cancer Model | In Vitro | In Vivo | IC50 In Vitro | Route of Administ Ration In Vivo | Dose Used In Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Didemmin B | Tunicate | Trididemnum sp. | Depsipeptide | Human permanent transformed cell lines HL-60, Daudi, Namalwa | ✓ | ✗ | 1 µM: 94%, 3.4%, 19.2% apoptosis, respectively | N/A | N/A | [75] |
| Human permanent | ✓ | ✗ | 1 µM: 14.4%, 6.7%, | N/A | N/A | |||||
| transformed cell lines BL-29, Naliaka, PDC-P1 | 7.9% | |||||||||
| Human | ✓ | ✗ | 1 µM: 18%, 3.3%, 90%, | N/A | N/A | |||||
| Permanent transformed cell | ||||||||||
| lines Molt-4, | 2% | |||||||||
| Jurkat, MM96, | apoptosis, | |||||||||
| and quiescent | respectively | |||||||||
| PBMC | ||||||||||
| Dehydrodidemnine B | Tunicate | Aplidium albicans | Depsipeptide | Murine Ehrlich ascitic mammary carcinoma cells and mouse model | ✓ | ✓ | <10 nM | i.p. | 2.5 mg | [76] |
| Tamandarin A and B | Tunicate | Unidentified. Didemnidae family | Depsipeptide | Human pancreatic carcinoma cell line BX-PC3 | ✓ | ✗ | <10 ng/mL | N/A | N/A | [77] |
| Human prostate cancer cell line DU-145 | ✓ | ✗ | <2.5 ng/mL | N/A | N/A | |||||
| Human head and neck carcinoma cell line UMSCC10b | ✓ | ✗ | <5 ng/mL | N/A | N/A | |||||
| Vitilevuamide | Tunicate | Didemnum cuculiferum | Bicyclic peptide | Human colon cancer cell line HCT-116 | ✓ | ✗ | 6 nM | N/A | N/A | [78] |
| Human lung adenocarcinoma cell line A549 | ✓ | ✗ | 124 nM | N/A | N/A | |||||
| Human melanoma cell line SK-MEL-5 | ✓ | ✗ | 311 nM | N/A | N/A | |||||
| Human kidney cancer cell line A498 | ✓ | ✗ | 311 nM | N/A | N/A | |||||
| Chinese hamster ovary cells | ✓ | ✗ | 3.1 µM | N/A | N/A | |||||
| Murine leukemia P338 allograft | ✗ | ✓ | N/A | i.p. | 6 –130 µg/Kg | |||||
| Diazonamide | Tunicate | Diazona angulata | Peptide | Human pancreatic cancer MIA PaCa-2, colon cancer HT-29, and MDA- MB-231-LM2 xenografts | ✗ | ✓ | N/A | i.v. | 2.25–36 mg/m2 | [79] |
| Trabectedin | Tunicate | Ecteinascidia turbinata | Alkaloid | In vivo cancer model. Not found | ✗ | ✓ | N/A | Not found | Not found | [80] |
| Eudistomin | Tunicate | Eudistoma olivaceum | Amino acid- derived alkaloid | Murine leukemia cell line L1210 and murine leukemia P388 allograft | ✓ | ✓ | 0.015–0.26µg/mL | i.p. | 0.03–8 mg/Kg | [81] |
| Lamellarin D | Tunicate | Lamellaria sp. | Amino acid- derived alkaloid | Human oralepidermoid carcinoma cell line KB | ✓ | ✗ | 0.04 µM | N/A | N/A | [82] |
| Human adenocarcinoma cell line A549 | ✓ | ✗ | 0.06 µM | N/A | N/A | |||||
| Multidrug resistant human small cell lung cancer cell line H69AR derived from NCI-H69 | ✓ | ✗ | 0.4 µM | N/A | N/A | |||||
| Human breast cancer cell line T47D | ✓ | ✗ | 0.00008 µM | N/A | N/A | |||||
| Triple negative breast cancer cell line MDA- MB-231 | ✓ | ✗ | 0.4 µM | N/A | N/A | |||||
| Human liver cancer cell line HuCCA-1 | ✓ | ✗ | 0.08 µM | N/A | N/A | |||||
| Human liver cancer cell line HepG2 | ✓ | ✗ | 0.02 µM | N/A | N/A | |||||
| Human liver cancer cell line S102 | ✓ | ✗ | 3.2 µM | N/A | N/A | |||||
| Human cervical epithelial carcinoma cell line HeLa | ✓ | ✗ | 0.06 µM | N/A | N/A | |||||
| Staurosporine | Tunicate | Eudistoma toealensis. | Indolocarbazole alkaloid | Murine leukemia cell line P388 | ✓ | ✗ | 0.1 µM | N/A | N/A | [82] |
| Human acute promyelocytic leukemia cell line HL-60 | ✓ | ✗ | 0.04 µM | N/A | N/A | |||||
| Human leukemia cell line CMK | ✓ | ✗ | 77% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line HL-60 | ✓ | ✗ | 72% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line JURKAT | ✓ | ✗ | 89% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line KASUMI-1 | ✓ | ✗ | 73% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line KG-1 | ✓ | ✗ | 72% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line LOUCY | ✓ | ✗ | 45% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line ML-2 | ✓ | ✗ | 69% growth inhibition at 2 mg/mL | N/A | N/A | [83] | ||||
| Human leukemia cell line MOLT-16 | ✓ | ✗ | 87% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line MONO-MAC-6 | ✓ | ✗ | 65% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line NB-4 | ✓ | ✗ | 75% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line PEER | ✓ | ✗ | 46% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Human leukemia cell line U-266 | ✓ | ✗ | 43% growth inhibition at 2 mg/mL | N/A | N/A | |||||
| Bistramide A | Tunicate | Lissoclinum bistratum | Cyclic polyether | Human oralepidermoid carcinoma cell line KB | ✓ | ✗ | 45 nM | N/A | N/A | [84] |
| Murine leukemia cell line P388 | ✓ | ✗ | 20 nM | N/A | N/A | |||||
| Mandelalide B and E | Tunicate | Lissoclinum sp. | Polyketide | Human non-small cell lung cancer cell line NCI-H460 | ✓ | ✗ | B: 25 nM, E: 2 µM | N/A | N/A | [85] |
| HeLa cells | ✓ | ✗ | B: 23 nM, E: 1.9 µM | N/A | N/A | |||||
| Human glioblastoma cell line U87-MG | ✓ | ✗ | B: 61 nM, E: >3 µM | N/A | N/A | |||||
| Human colon carcinoma cell line HCT-116 | ✓ | ✗ | B: 54 nM, E: >3 µM | N/A | N/A |
| Compound Name | Marine Organism | Species Name | Active Derivative | Cancer Model | I n | I n | IC50 In Vitro | Route of Administration In Vivo | Dose Used In Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Jorumycin | Mollusk | Jorunna funebris | Isoquinoline alkaloid | Murine leukemia cell line P388 | ✓ | ✗ | 12.5 ng/mL | N/A | N/A | [86] |
| Human lung adenocarcinoma cell line A549 | ✓ | ✗ | 12.5 ng/mL | N/A | N/A | |||||
| Human colon cancer cell line HT29 | ✓ | ✗ | 12.5 ng/mL | N/A | N/A | |||||
| Human melanoma cell line MEL28 | ✓ | ✗ | 12.5 ng/mL | N/A | N/A | |||||
| Human colon cancer cell line HCT116 | ✓ | ✗ | 0.57 nM | N/A | N/A | [87] | ||||
| Human lung cancer cell line QC56 | ✓ | ✗ | 0.76 nM | N/A | N/A | |||||
| Human prostate cancer cell line DU145 | ✓ | ✗ | 0.49 nM | N/A | N/A | |||||
| Dolastatin 10 | Mollusk | Dolabella auricularia | Linear pentapeptide | Murine leukemia cell line P338 | ✓ | ✗ | 4.6 × 10−5 ng/mL | N/A | N/A | [88] |
| Human ovarian cancer cell line OVCAR-3 | ✓ | ✗ | 9.5 × 10−7 µg/mL | N/A | N/A | |||||
| Human glioma cell line SF-295 | ✓ | ✗ | 7.6 × 10−6 µg/mL | N/A | N/A | |||||
| Human kidney cancer cell line A498 | ✓ | ✗ | 2.6 × 10−5 µg/mL | N/A | N/A | |||||
| Human non-small cell lung cancer cell line NCI-H460 | ✓ | ✗ | 3.4 × 10−6 µg/mL | N/A | N/A | |||||
| Human colon cancer cell line KM20L2 | ✓ | ✗ | 4.7 × 10−6 µg/mL | N/A | N/A | |||||
| Human melanoma cell line SK-MEL-5 | ✓ | ✗ | 7.4 × 10−6 µg/mL | N/A | N/A | |||||
| Human colon adenocarcinoma cell line LoVo | ✓ | ✗ | 0.052 nM | N/A | N/A | [16] | ||||
| HeLa-derived cell line KB | ✓ | ✗ | 0.076 nM | N/A | N/A | |||||
| Dolastatin 15 | Mollusk | Dolabella auricularia | Linear pentapeptide | Human multiple myeloma cell lines RPMI8226, U266, and IM9 | ✓ | ✗ | 0.5–1 nM | N/A | N/A | [89] |
| Kahalalide F | Mollusk | Elysia rufescens | Depsipeptide | Human breast cancer cell line H5578T and Hs-578T | ✓ | ✗ | 0.162 and 0.479 µM | N/A | N/A | [90] |
| Human lung adenocarcinoma cell line A549 | ✓ | ✗ | 0.135 µM | N/A | N/A | |||||
| Human colon cancer cell line (not specified) | ✓ | ✗ | 0.162–0.288 µM | N/A | N/A | |||||
| Elisidepsin (KF synthetic derivative) | Mollusk | Elysia rufescens | Depsipeptide | Human breast cancer cell lines ZR-75-1, SKBR3, MDA-MB-361, MDA- MB-231, and MCF7 | ✓ | ✗ | 0.40, 0.5, 1.25, 4.7, and 8 µM, respectively | N/A | N/A | [91] |
| Human colon cancer cell lines Colo205, HCC2998, HT29, Colo205R, and HCT116 | ✓ | ✗ | 0.75, 1.2, 3.7, 6.1, and 7.2 µM, respectively | N/A | N/A | |||||
| Human head and neck cancer cell lines SQ20B, HEP2, and SCC61 | ✓ | ✗ | 3.5, 4.3, and 5.6 µM, respectively | N/A | N/A | |||||
| Human hepatocarcinoma cell line SK-HEP1 | ✓ | ✗ | 6 µM | N/A | N/A | |||||
| Human lung cancer cell lines HOP62 and HOP92 | ✓ | ✗ | 6.3 and 8 µM | N/A | N/A | |||||
| Human melanoma cell line MDA-MB-435 | ✓ | ✗ | 4.4 µM | N/A | N/A | |||||
| Human ovarian cancer cell lines IGROV1 and OVCAR3 | ✓ | ✗ | 4.2 and 7.3 µM | N/A | N/A | |||||
| Human pancreatic cancer cell lines CAPAN1 and MiaCaPa2 | ✓ | ✗ | 5 and 8.8 µM | N/A | N/A | |||||
| Human prostate cancer cell lines DU145 and PC3 | ✓ | ✗ | 1.26 and 1.8 µM | N/A | N/A | |||||
| Kulokekahilide-2 | Mollusk | Philinopsis speciosa | Depsipeptide | Murine leukemia cell lines P338 | ✓ | ✗ | 4.2 nM | N/A | N/A | [92] |
| Human ovarian cancer cell line SK-OV-3 | ✓ | ✗ | 7.5 nM | N/A | N/A | |||||
| Human melanoma cell line MDA-MB-435 | ✓ | ✗ | 14.6 nM | N/A | N/A |
| Compound Name | Marine Organism | Species Name | Active Derivative | Cancer Model | In Vitro | In Vivo | IC50 in Vitro | Route of Administration In Vivo | Dose Used In Vivo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Tambjamine K | Bryozoan | Virididentula dentate | Bipyrrolic alkaloid | Human colon cancer cell line HCT116 | ✓ | ✗ | Cp 12: 13.7 µM, Cp 13: 3.6 µM, Cp 14: 0.14 µM | N/A | N/A | [93] |
| Human breast cancer cell line MDA-MB-231 | ✓ | ✗ | Cp 12: 15.3 µM, Cp 13: 3.5 µM | N/A | N/A | |||||
| Indole-based Tambjamine analogs | Bryozoan | Virididentula dentate. | Alkaloid | Human lung adenocarcinoma cell line A549 | ✓ | ✗ | Cp 1: 10.66 µM and Cp 2: 7.61 µM | N/A | N/A | [94] |
| Human small cell lung carcinoma cell line DMS53 and xenograft | ✓ | ✓ | Cp 1: 8.04 µM and Cp 2: 6.46 µM | i.p. | 6 mg/Kg | |||||
| Human lung squamous carcinoma cell line SW900 | ✓ | ✗ | Cp 1: 8.67 µM and Cp 2: 7.55 µM | N/A | N/A | |||||
| Human large cell lung cancer cell line H460 | ✓ | ✗ | Cp 1: 8.37 µM and Cp 2: 7.29 µM | N/A | N/A | |||||
| Human lung cancer primary culture #8 | ✓ | ✗ | Cp 1: 4.04 µM and Cp 2: 3.34 µM | N/A | N/A | |||||
| Human lung cancer primary culture #13 | ✓ | ✗ | Cp 1: 4.34 µM and Cp 2: 4.03 µM | N/A | N/A | |||||
| Bryostatin 1 | Bryozoan | Bugula neritina | Macrocyclic lactone | Murine leukemia cell line P388 | ✓ | ✗ | 0.25 nM | N/A | N/A | [95] |
| Bryostatin 5 and 8 | Bryozoan | Bugula neritina. | Macrocyclic lactone | Murine melanoma K1735-M2 allograft | ✗ | ✓ | N/A | i.p. | 1 µg | [96] |
| Compound Name | Commercial Name | Marine Organism | Active Derivative | Molecular Target | Cancer Type | Year of 1st Approval and Agency or Clinical Phase |
|---|---|---|---|---|---|---|
| Cytarabine (Ara-C) | Cytosar-U® Depocyt® | Sponge | Nucleoside | DNA polymerase | Acute myeloid leukemia, non-Hodgkin’s lymphoma | 196 9FDA |
| Fludarabine | Fludara® | Sponge | Nucleoside | DNA polymerase | Chronic lymphocytic leukemia, and indolent B-cell lymphoma | 2008 FDA |
| Nelarabine (506U78) | Arranon® | Sponge | Nucleoside | DNA polymerase | T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma | 2005 FDA |
| Trabectedin (ET-743) | Yondelis® | Tunicate | Alkaloid | Minor groove of DNA | Soft tissue sarcoma, ovarian cancer | 2007 EMEA |
| Eribulin mesylate (E7389) | Halaven® | Sponge | Polyketide | Microtubule | Locally advanced or metastatic breast cancer | 2010 FDA |
| Brentuximab vedotin (SGN-35) | Adcetris® | Mollusk and cyanobacteria | ADC (anti CD30-MMAE) | CD30 and microtubules | Anaplastic large T-cell malignant lymphoma, Hodgkin’s lymphoma | 2011 FDA |
| Plitidepsin | Aplidin® | Tunicate | Cyclic depsi-peptide | Rac1 and JNK activation | Multiple myeloma, T-cell lymphoma, leukemia | 2018 ATGA |
| Polatuzumab vedotin (DCDS-4501A) | Polivy® | Mollusk and cyanobacteria | ADC (anti CD79b-MMAE) | CD79b and microtubules | Diffuse large B-cell lymphoma | 201 9FDA |
| Plinabulin (NPI-2358) | NA | Fungi | Amide | Microtubules and JNK | Non-small cell lung cancer | III |
| Lurbinectedin (PM01183) | NA | Synthetic form from tunicate | Alkaloid | Minor groove of DNA | Small cell lung cancer Ovarian cancer | III |
| Depatuxizumab mafodotin (ABT-414) | NA | Mollusk and cyanobacteria | ADC (anti EGFR-MMAF) | EGFR and microtubule | Glioblastoma multiforme | III |
| Enfortumab vedotin (ASG-22ME) | NA | Mollusk and cyanobacteria | ADC (anti Nectin-4-MMAE) | Nectin-4 and microtubule | Urothelial cancer | III |
| Marizomib (NPI-0052) | NA | Bacteria | Beta-lactone | 20S proteasome | Glioblastoma | III |
| Limitations | Strategies to Overcome limitations | References |
|---|---|---|
| Lack of sustainable supply | Increase development of synthetic or hemi-synthetic derivatives from the biological source | [108,169] |
| Low production of bioactive compounds | Changing culture conditions, genetic engineering of organisms | [37,178,185] |
| Properly designed and implemented extraction methodologies | [179] | |
| Structural complexity of the marine compounds | In silico screening programs, NMR and MS | [75,185,186,187,188] |
| Correct taxonomic determination | Genomic approaches | [191,192,193,194] |
| Moderated efficacy | Conjugation with antibodies | [196] |
| Encapsulation with nanoparticles | [201,202] | |
| Combination with other drugs | [201,202] | |
| Use of cell-penetrating peptides and tumor homing peptides | [197,199,200,203] | |
| High market value | Rigorous planning on the usage of marine-derived drugs | [162] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, E.; Sorolla, M.A.; Gopal Krishnan, P.D.; Sorolla, A. From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules 2020, 10, 248. https://doi.org/10.3390/biom10020248
Wang E, Sorolla MA, Gopal Krishnan PD, Sorolla A. From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules. 2020; 10(2):248. https://doi.org/10.3390/biom10020248
Chicago/Turabian StyleWang, Edina, Maria Alba Sorolla, Priya Darshini Gopal Krishnan, and Anabel Sorolla. 2020. "From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds" Biomolecules 10, no. 2: 248. https://doi.org/10.3390/biom10020248
APA StyleWang, E., Sorolla, M. A., Gopal Krishnan, P. D., & Sorolla, A. (2020). From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules, 10(2), 248. https://doi.org/10.3390/biom10020248