New Insights into ADAMTS Metalloproteases in the Central Nervous System

Abstract
:1. Introduction
2. Lecticans in the CNS
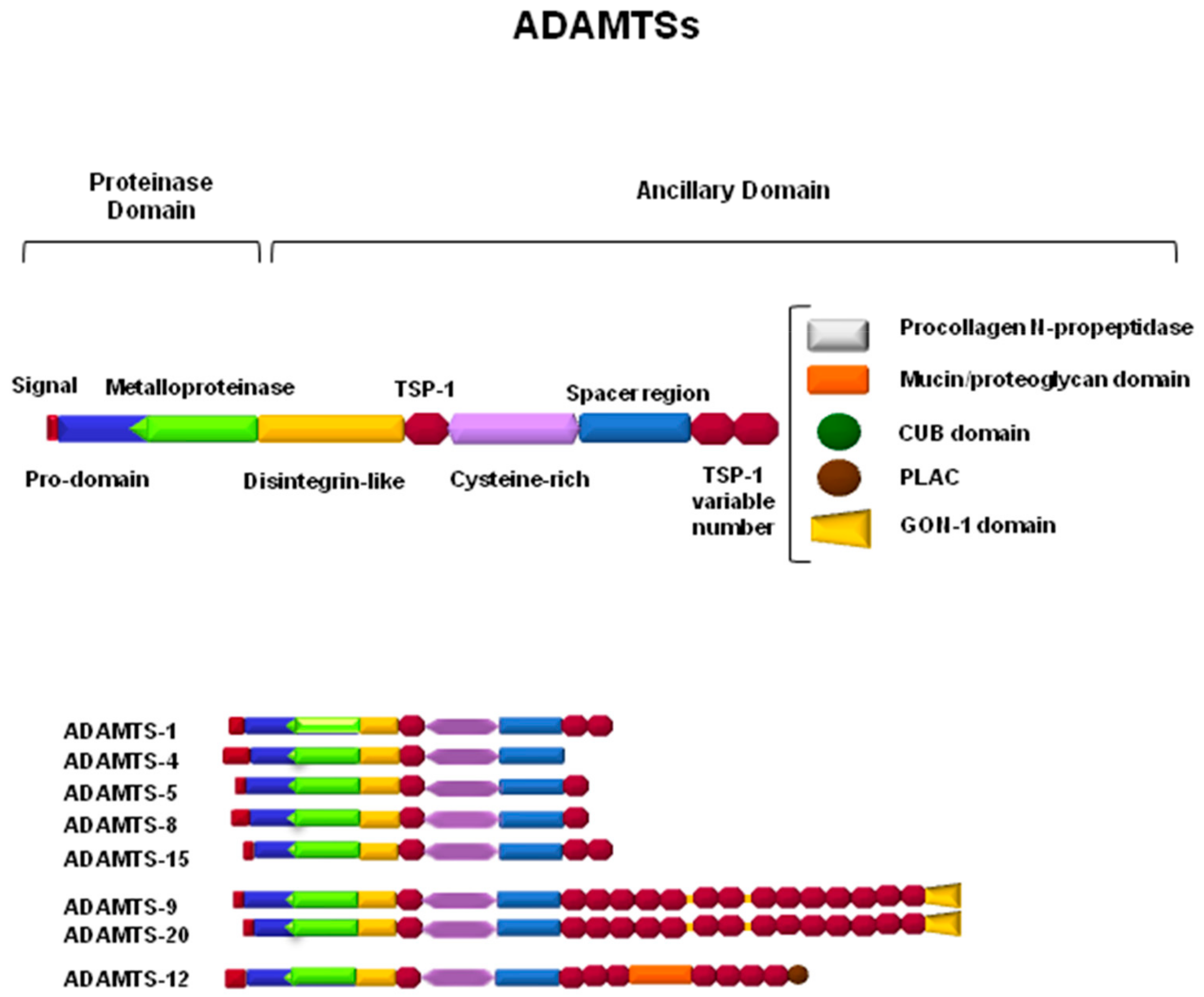
3. ADAMTSs in the CNS
4. ADAMTS Functions in Normal and Pathological CNS
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodeling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, M.; Block, T.J.; Rakian, R.; Li, Q.; Wang, E.; Reilly, M.A.; Dean, D.D.; Chen, X.D. One size does not fit all: Developing a cell-specific niche for in vitro study of cell behavior. Matrix Biol. 2016, 52–54, 426–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Overall, C.M. Protease degradomics: A new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002, 3, 509–519. [Google Scholar] [CrossRef]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 2016, 128, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Fontanil, T.; Mohamedi, Y.; Cobo, T.; Cal, S.; Obaya, A.J. Novel associations within the tumor microenvironment: fibulins meet ADAMTSs. Front. Oncol. 2019, 9, 796. [Google Scholar] [CrossRef]
- Fontanil, T.; Mohamedi, Y.; Moncada-Pazos, A.; Cobo, T.; Vega, J.A.; Cobo, J.L.; Garcia-Suarez, O.; Cobo, J.; Obaya, A.J.; Cal, S. Neurocan is a new substrate for the ADAMTS12 metalloprotease: potential implications in neuropathies. Cell. Physiol. Biochem. 2019, 52, 1003–1016. [Google Scholar] [CrossRef]
- McRae, P.A.; Porter, B.E. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem. Int. 2012, 61, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.W.; Reichert, J.R.; Davis, C.J.; Harding, J.W. Neural plasticity and the brain renin-angiotensin system. Neurosci. Biobehav. Rev. 2002, 26, 529–552. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M. Extracellular matrix molecules and synaptic plasticity. Nat. Rev. Neurosci. 2003, 4, 456–468. [Google Scholar] [CrossRef]
- Sandvig, A.; Berry, M.; Barrett, L.B.; Butt, A.; Logan, A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: Expression, receptor signaling, and correlation with axon regeneration. Glia 2004, 46, 225–251. [Google Scholar] [CrossRef] [PubMed]
- Barros, C.S.; Franco, S.J.; Muller, U. Extracellular matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Freeman, C.; Jacobson, G.A.; Small, D.H. Proteoglycans in the central nervous system: Role in development, neural repair, and Alzheimer’s disease. IUBMB Life 2013, 65, 108–120. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Geller, H.M. Extracellular matrix and traumatic brain injury. J. Neurosci. Res. 2018, 96, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.M.; Hsieh-Wilson, L.C. Sugar-dependent modulation of neuronal development, regeneration, and plasticity by chondroitin sulfate proteoglycans. Exp. Neurol. 2015, 274, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Myer, D.J.; Gurkoff, G.G.; Lee, S.M.; Hovda, D.A.; Sofroniew, M.V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006, 129, 2761–2772. [Google Scholar] [CrossRef]
- Siebert, J.R.; Conta Steencken, A.; Osterhout, D.J. Chondroitin sulfate proteoglycans in the nervous system: Inhibitors to repair. BioMed Res. Int. 2014, 2014, 845323. [Google Scholar] [CrossRef] [Green Version]
- McKeon, R.J.; Hoke, A.; Silver, J. Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp. Neurol. 1995, 136, 32–43. [Google Scholar] [CrossRef]
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000, 80, 1267–1290. [Google Scholar] [CrossRef] [PubMed]
- Nandadasa, S.; Foulcer, S.; Apte, S.S. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014, 35, 34–41. [Google Scholar] [CrossRef]
- Kischel, P.; Waltregny, D.; Dumont, B.; Turtoi, A.; Greffe, Y.; Kirsch, S.; De Pauw, E.; Castronovo, V. Versican overexpression in human breast cancer lesions: known and new isoforms for stromal tumor targeting. Int. J. Cancer 2010, 126, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Oohashi, T.; Edamatsu, M.; Bekku, Y.; Carulli, D. The hyaluronan and proteoglycan link proteins: Organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp. Neurol. 2015, 274, 134–144. [Google Scholar] [CrossRef]
- Maeda, N. Structural variation of chondroitin sulfate and its roles in the central nervous system. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, S.; Andersen, J.S.; Maurel, P.; Margolis, R.U. Localization of aggrecan and versican in the developing rat central nervous system. Dev. Dyn. 2003, 227, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Kelly, G.M.; Zerillo, C.A.; Gray, G.; Tiemeyer, M.; Hockfield, S. Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J. Neurosci. 2002, 22, 7536–7547. [Google Scholar] [CrossRef]
- Rittenhouse, E.; Dunn, L.C.; Cookingham, J.; Calo, C.; Spiegelman, M.; Dooher, G.B.; Bennett, D. Cartilage matrix deficiency (cmd): A new autosomal recessive lethal mutation in the mouse. J. Embryol. Exp. Morphol. 1978, 43, 71–84. [Google Scholar]
- Giamanco, K.A.; Morawski, M.; Matthews, R.T. Perineuronal net formation and structure in aggrecan knockout mice. Neuroscience 2010, 170, 1314–1327. [Google Scholar] [CrossRef]
- Rowlands, D.; Lensjo, K.K.; Dinh, T.; Yang, S.; Andrews, M.R.; Hafting, T.; Fyhn, M.; Fawcett, J.W.; Dick, G. Aggrecan directs extracellular matrix-mediated neuronal plasticity. J. Neurosci. 2018, 38, 10102–10113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, R.A.; Morgenstern, D.A.; Shearer, M.C.; Adcock, K.H.; Pesheva, P.; Fawcett, J.W. Versican is upregulated in CNS injury and is a product of oligodendrocyte lineage cells. J. Neurosci. 2002, 22, 2225–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oohashi, T.; Hirakawa, S.; Bekku, Y.; Rauch, U.; Zimmermann, D.R.; Su, W.D.; Ohtsuka, A.; Murakami, T.; Ninomiya, Y. Bral1, a brain-specific link protein, colocalizing with the versican V2 isoform at the nodes of Ranvier in developing and adult mouse central nervous systems. Mol. Cell. Neurosci. 2002, 19, 43–57. [Google Scholar] [CrossRef]
- Gu, W.L.; Fu, S.L.; Wang, Y.X.; Li, Y.; Wang, X.F.; Xu, X.M.; Lu, P.H. Expression and regulation of versican in neural precursor cells and their lineages. Acta Pharmacol. Sin. 2007, 28, 1519–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaudry, D.; Chen, Y.; Hsu, C.M.; Eiden, L.E. PC12 cells as a model to study the neurotrophic activities of PACAP. Ann. N. Y. Acad. Sci. 2002, 971, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sheng, W.; Chen, L.; Dong, H.; Lee, V.; Lu, F.; Wong, C.S.; Lu, W.Y.; Yang, B.B. Versican V1 isoform induces neuronal differentiation and promotes neurite outgrowth. Mol. Biol. Cell 2004, 15, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Dours-Zimmermann, M.T.; Maurer, K.; Rauch, U.; Stoffel, W.; Fassler, R.; Zimmermann, D.R. Versican V2 assembles the extracellular matrix surrounding the nodes of ranvier in the CNS. J. Neurosci. 2009, 29, 7731–7742. [Google Scholar] [CrossRef] [Green Version]
- Mjaatvedt, C.H.; Yamamura, H.; Capehart, A.A.; Turner, D.; Markwald, R.R. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev. Biol. 1998, 202, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Brakebusch, C.; Seidenbecher, C.I.; Asztely, F.; Rauch, U.; Matthies, H.; Meyer, H.; Krug, M.; Bockers, T.M.; Zhou, X.; Kreutz, M.R.; et al. Brevican-deficient mice display impaired hippocampal CA1 long-term potentiation but show no obvious deficits in learning and memory. Mol. Cell. Biol. 2002, 22, 7417–7427. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Hagihara, K.; Suzuki, M.; Yamaguchi, Y. Brevican in the developing hippocampal fimbria: differential expression in myelinating oligodendrocytes and adult astrocytes suggests a dual role for brevican in central nervous system fiber tract development. J. Comp. Neurol. 2001, 432, 285–295. [Google Scholar] [CrossRef]
- Sonntag, M.; Blosa, M.; Schmidt, S.; Reimann, K.; Blum, K.; Eckrich, T.; Seeger, G.; Hecker, D.; Schick, B.; Arendt, T.; et al. Synaptic coupling of inner ear sensory cells is controlled by brevican-based extracellular matrix baskets resembling perineuronal nets. BMC Biol. 2018, 16, 99. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, K.L.; Xu, X.; Ogawa, Y.; Frischknecht, R.; Seidenbecher, C.I.; Shrager, P.; Rasband, M.N. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J. Cell Biol. 2007, 178, 875–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischknecht, R.; Seidenbecher, C.I. Brevican: A key proteoglycan in the perisynaptic extracellular matrix of the brain. Int. J. Biochem. Cell Biol. 2012, 44, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.H.; Brakebusch, C.; Matthies, H.; Oohashi, T.; Hirsch, E.; Moser, M.; Krug, M.; Seidenbecher, C.I.; Boeckers, T.M.; Rauch, U.; et al. Neurocan is dispensable for brain development. Mol. Cell. Biol. 2001, 21, 5970–5978. [Google Scholar] [CrossRef] [Green Version]
- Quaglia, X.; Beggah, A.T.; Seidenbecher, C.; Zurn, A.D. Delayed priming promotes CNS regeneration post-rhizotomy in neurocan and brevican-deficient mice. Brain 2008, 131, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschling, C.; Wegrzyn, D.; Denecke, B.; Faissner, A. Elimination of the four extracellular matrix molecules tenascin-C, tenascin-R, brevican and neurocan alters the ratio of excitatory and inhibitory synapses. Sci. Rep. 2019, 9, 13939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, S.E.; Li, J.; Schauwecker, P.E.; McNeill, T.H.; Salton, S.R. Comparison of RPTP zeta/beta, phosphacan, and trkB mRNA expression in the developing and adult rat nervous system and induction of RPTP zeta/beta and phosphacan mRNA following brain injury. Brain Res. Mol. Brain Res. 1996, 40, 79–96. [Google Scholar] [CrossRef]
- Harroch, S.; Palmeri, M.; Rosenbluth, J.; Custer, A.; Okigaki, M.; Shrager, P.; Blum, M.; Buxbaum, J.D.; Schlessinger, J. No obvious abnormality in mice deficient in receptor protein tyrosine phosphatase beta. Mol. Cell. Biol. 2000, 20, 7706–7715. [Google Scholar] [CrossRef] [Green Version]
- Faissner, A.; Clement, A.; Lochter, A.; Streit, A.; Mandl, C.; Schachner, M. Isolation of a neural chondroitin sulfate proteoglycan with neurite outgrowth promoting properties. J. Cell Biol. 1994, 126, 783–799. [Google Scholar] [CrossRef]
- Garwood, J.; Schnadelbach, O.; Clement, A.; Schutte, K.; Bach, A.; Faissner, A. DSD-1-proteoglycan is the mouse homolog of phosphacan and displays opposing effects on neurite outgrowth dependent on neuronal lineage. J. Neurosci. 1999, 19, 3888–3899. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N. Proteoglycans and neuronal migration in the cerebral cortex during development and disease. Front. Neurosci. 2015, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inatani, M.; Honjo, M.; Otori, Y.; Oohira, A.; Kido, N.; Tano, Y.; Honda, Y.; Tanihara, H. Inhibitory effects of neurocan and phosphacan on neurite outgrowth from retinal ganglion cells in culture. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1930–1938. [Google Scholar]
- Theocharidis, U.; Long, K.; Ffrench-Constant, C.; Faissner, A. Regulation of the neural stem cell compartment by extracellular matrix constituents. Prog. Brain Res. 2014, 214, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Negron-Oyarzo, I.; Lara-Vasquez, A.; Palacios-Garcia, I.; Fuentealba, P.; Aboitiz, F. Schizophrenia and reelin: a model based on prenatal stress to study epigenetics, brain development and behavior. Biol. Res. 2016, 49, 16. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, N.C.; Anderson, R.C.; McDermott, K.W. Reelin: Diverse roles in central nervous system development, health and disease. Int. J. Biochem. Cell Biol. 2019, 112, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, N.J.; Trossbach, S.V.; Kober, S.; Walter, S.; Prikulis, I.; Weggen, S.; Korth, C. Disrupted in Schizophrenia 1 regulates the processing of reelin in the perinatal cortex. Schizophr. Res. 2017, 215, 506–513. [Google Scholar] [CrossRef]
- Won, S.J.; Kim, S.H.; Xie, L.; Wang, Y.; Mao, X.O.; Jin, K.; Greenberg, D.A. Reelin-deficient mice show impaired neurogenesis and increased stroke size. Exp. Neurol. 2006, 198, 250–259. [Google Scholar] [CrossRef]
- Jossin, Y.; Gui, L.; Goffinet, A.M. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J. Neurosci. 2007, 27, 4243–4252. [Google Scholar] [CrossRef] [Green Version]
- Lemarchant, S. Relevance of the proteolytic processing of Reelin by ADAMTS-3 in brain functions. J. Neurosci. 2017, 37, 6814–6815. [Google Scholar] [CrossRef] [Green Version]
- Porter, S.; Clark, I.M.; Kevorkian, L.; Edwards, D.R. The ADAMTS metalloproteinases. Biochem. J. 2005, 386, 15–27. [Google Scholar] [CrossRef]
- Dancevic, C.M.; McCulloch, D.R.; Ward, A.C. The ADAMTS hyalectanase family: Biological insights from diverse species. Biochem. J. 2016, 473, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Mead, T.J.; Apte, S.S. ADAMTS proteins in human disorders. Matrix Biol. 2018, 71–72, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Cal, S.; Obaya, A.J.; Llamazares, M.; Garabaya, C.; Quesada, V.; Lopez-Otin, C. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene 2002, 283, 49–62. [Google Scholar] [CrossRef]
- Gurses, M.S.; Ural, M.N.; Gulec, M.A.; Akyol, O.; Akyol, S. Pathophysiological function of ADAMTS enzymes on molecular mechanism of Alzheimer’s Disease. Aging Dis. 2016, 7, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llamazares, M.; Cal, S.; Quesada, V.; Lopez-Otin, C. Identification and characterization of ADAMTS-20 defines a novel subfamily of metalloproteinases-disintegrins with multiple thrombospondin-1 repeats and a unique GON domain. J. Biol. Chem. 2003, 278, 13382–13389. [Google Scholar] [CrossRef] [Green Version]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [Green Version]
- Stanton, H.; Melrose, J.; Little, C.B.; Fosang, A.J. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim. Biophys. Acta 2011, 1812, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Colige, A.; Vandenberghe, I.; Thiry, M.; Lambert, C.A.; Van Beeumen, J.; Li, S.W.; Prockop, D.J.; Lapiere, C.M.; Nusgens, B.V. Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3. J. Biol. Chem. 2002, 277, 5756–5766. [Google Scholar] [CrossRef] [Green Version]
- Perez-Garcia, S.; Carrion, M.; Villanueva-Romero, R.; Hermida-Gomez, T.; Fernandez-Moreno, M.; Mellado, M.; Blanco, F.J.; Juarranz, Y.; Gomariz, R.P. Wnt and RUNX2 mediate cartilage breakdown by osteoarthritis synovial fibroblast-derived ADAMTS-7 and -12. J. Cell. Mol. Med. 2019, 23, 3974–3983. [Google Scholar] [CrossRef]
- Fujikawa, K.; Suzuki, H.; McMullen, B.; Chung, D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood 2001, 98, 1662–1666. [Google Scholar] [CrossRef]
- Westling, J.; Fosang, A.J.; Last, K.; Thompson, V.P.; Tomkinson, K.N.; Hebert, T.; McDonagh, T.; Collins-Racie, L.A.; LaVallie, E.R.; Morris, E.A.; et al. ADAMTS4 cleaves at the aggrecanase site (Glu373-Ala374) and secondarily at the matrix metalloproteinase site (Asn341-Phe342) in the aggrecan interglobular domain. J. Biol. Chem. 2002, 277, 16059–16066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, P.; Dalal, K. ADAMTS-4 and ADAMTS-5: Key enzymes in osteoarthritis. J. Cell. Biochem. 2011, 112, 3507–3514. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.; Pratta, M.; Liu, R.Q.; Abbaszade, I.; Ross, H.; Burn, T.; Arner, E. The thrombospondin motif of aggrecanase-1 (ADAMTS-4) is critical for aggrecan substrate recognition and cleavage. J. Biol. Chem. 2000, 275, 25791–25797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, E.; Yan, X.; Zhang, M.; Chang, X.; Bai, Z.; He, Y.; Yuan, Z. Aggrecanases in the human synovial fluid at different stages of osteoarthritis. Clin. Rheumatol. 2013, 32, 797–803. [Google Scholar] [CrossRef]
- Cross, N.A.; Chandrasekharan, S.; Jokonya, N.; Fowles, A.; Hamdy, F.C.; Buttle, D.J.; Eaton, C.L. The expression and regulation of ADAMTS-1, -4, -5, -9, and -15, and TIMP-3 by TGFbeta1 in prostate cells: relevance to the accumulation of versican. Prostate 2005, 63, 269–275. [Google Scholar] [CrossRef]
- Fu, Y.; Nagy, J.A.; Brown, L.F.; Shih, S.C.; Johnson, P.Y.; Chan, C.K.; Dvorak, H.F.; Wight, T.N. Proteolytic cleavage of versican and involvement of ADAMTS-1 in VEGF-A/VPF-induced pathological angiogenesis. J. Histochem. Cytochem. 2011, 59, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.L.; Hou, L.; Somerville, R.; Young, M.E.; Apte, S.S.; Pavan, W.J. The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet. 2008, 4, e1000003. [Google Scholar] [CrossRef] [Green Version]
- Kenagy, R.D.; Plaas, A.H.; Wight, T.N. Versican degradation and vascular disease. Trends Cardiovasc. Med. 2006, 16, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Gary, S.C.; Kelly, G.M.; Hockfield, S. BEHAB/brevican: A brain-specific lectican implicated in gliomas and glial cell motility. Curr. Opin. Neurobiol. 1998, 8, 576–581. [Google Scholar] [CrossRef]
- Nakada, M.; Miyamori, H.; Kita, D.; Takahashi, T.; Yamashita, J.; Sato, H.; Miura, R.; Yamaguchi, Y.; Okada, Y. Human glioblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol. 2005, 110, 239–246. [Google Scholar] [CrossRef]
- Yuan, W.; Matthews, R.T.; Sandy, J.D.; Gottschall, P.E. Association between protease-specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate-treated rats. Neuroscience 2002, 114, 1091–1101. [Google Scholar] [CrossRef]
- Hisanaga, A.; Morishita, S.; Suzuki, K.; Sasaki, K.; Koie, M.; Kohno, T.; Hattori, M. A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4) cleaves Reelin in an isoform-dependent manner. FEBS Lett. 2012, 586, 3349–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstic, D.; Rodriguez, M.; Knuesel, I. Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS ONE 2012, 7, e47793. [Google Scholar] [CrossRef] [PubMed]
- Jungers, K.A.; Le Goff, C.; Somerville, R.P.; Apte, S.S. Adamts9 is widely expressed during mouse embryo development. Gene Expr. Patterns 2005, 5, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.K.; Haddock, G.; Surr, J.; Plumb, J.; Bunning, R.A.; Buttle, D.J.; Woodroofe, M.N. Differential expression of ADAMTS-1, -4, -5 and TIMP-3 in rat spinal cord at different stages of acute experimental autoimmune encephalomyelitis. J. Autoimmun. 2006, 26, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Ajmo, J.M.; Eakin, A.K.; Hamel, M.G.; Gottschall, P.E. Discordant localization of WFA reactivity and brevican/ADAMTS-derived fragment in rodent brain. BMC Neurosci. 2008, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Miguel, R.F.; Pollak, A.; Lubec, G. Metalloproteinase ADAMTS-1 but not ADAMTS-5 is manifold overexpressed in neurodegenerative disorders as Down syndrome, Alzheimer’s and Pick’s disease. Brain Res. Mol. Brain Res. 2005, 133, 1–5. [Google Scholar] [CrossRef]
- Lemarchant, S.; Wojciechowski, S.; Vivien, D.; Koistinaho, J. ADAMTS-4 in central nervous system pathologies. J. Neurosci. Res. 2017, 95, 1703–1711. [Google Scholar] [CrossRef]
- Lemarchant, S.; Pomeshchik, Y.; Kidin, I.; Karkkainen, V.; Valonen, P.; Lehtonen, S.; Goldsteins, G.; Malm, T.; Kanninen, K.; Koistinaho, J. ADAMTS-4 promotes neurodegeneration in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2016, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Lemarchant, S.; Pruvost, M.; Montaner, J.; Emery, E.; Vivien, D.; Kanninen, K.; Koistinaho, J. ADAMTS proteoglycanases in the physiological and pathological central nervous system. J. Neuroinflamm. 2013, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Hamel, M.G.; Mayer, J.; Gottschall, P.E. Altered production and proteolytic processing of brevican by transforming growth factor beta in cultured astrocytes. J. Neurochem. 2005, 93, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, R.; Imagama, S.; Natori, T.; Ohgomori, T.; Muramoto, A.; Shinjo, R.; Matsuyama, Y.; Ishiguro, N.; Kadomatsu, K. The endogenous proteoglycan-degrading enzyme ADAMTS-4 promotes functional recovery after spinal cord injury. J. Neuroinflamm. 2012, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.K.; Haddock, G.; Stock, C.J.; Allan, S.; Surr, J.; Bunning, R.A.; Buttle, D.J.; Woodroofe, M.N. ADAMTS-1 and -4 are up-regulated following transient middle cerebral artery occlusion in the rat and their expression is modulated by TNF in cultured astrocytes. Brain Res. 2006, 1088, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Levy, C.; Brooks, J.M.; Chen, J.; Su, J.; Fox, M.A. Cell-specific and developmental expression of lectican-cleaving proteases in mouse hippocampus and neocortex. J. Comp. Neurol. 2015, 523, 629–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, S.N.; Iruela-Arispe, M.L. Expression of ADAMTS1 during murine development. Mech. Dev. 2002, 115, 181–185. [Google Scholar] [CrossRef]
- Gunther, W.; Skaftnesmo, K.O.; Arnold, H.; Bjerkvig, R.; Terzis, A.J. Distribution patterns of the anti-angiogenic protein ADAMTS-1 during rat development. Acta Histochem. 2005, 107, 121–131. [Google Scholar] [CrossRef]
- Sasaki, M.; Seo-Kiryu, S.; Kato, R.; Kita, S.; Kiyama, H. A disintegrin and metalloprotease with thrombospondin type1 motifs (ADAMTS-1) and IL-1 receptor type 1 mRNAs are simultaneously induced in nerve injured motor neurons. Brain Res. Mol. Brain Res. 2001, 89, 158–163. [Google Scholar] [CrossRef]
- Gottschall, P.E.; Howell, M.D. ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 2015, 44–46, 70–76. [Google Scholar] [CrossRef]
- Satoh, K.; Suzuki, N.; Yokota, H. ADAMTS-4 (a disintegrin and metalloproteinase with thrombospondin motifs) is transcriptionally induced in beta-amyloid treated rat astrocytes. Neurosci. Lett. 2000, 289, 177–180. [Google Scholar] [CrossRef]
- Lemarchant, S.; Dunghana, H.; Pomeshchik, Y.; Leinonen, H.; Kolosowska, N.; Korhonen, P.; Kanninen, K.M.; Garcia-Berrocoso, T.; Montaner, J.; Malm, T.; et al. Anti-inflammatory effects of ADAMTS-4 in a mouse model of ischemic stroke. Glia 2016, 64, 1492–1507. [Google Scholar] [CrossRef]
- Lemarchant, S.; Pruvost, M.; Hebert, M.; Gauberti, M.; Hommet, Y.; Briens, A.; Maubert, E.; Gueye, Y.; Feron, F.; Petite, D.; et al. tPA promotes ADAMTS-4-induced CSPG degradation, thereby enhancing neuroplasticity following spinal cord injury. Neurobiol. Dis. 2014, 66, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Moncada-Pazos, A.; Obaya, A.J.; Llamazares, M.; Heljasvaara, R.; Suarez, M.F.; Colado, E.; Noel, A.; Cal, S.; Lopez-Otin, C. ADAMTS-12 metalloprotease is necessary for normal inflammatory response. J. Biol. Chem. 2012, 287, 39554–39563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawcett, J.W. The extracellular matrix in plasticity and regeneration after CNS injury and neurodegenerative disease. Prog. Brain Res. 2015, 218, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Gottschall, P.E. Lectican proteoglycans, their cleaving metalloproteinases, and plasticity in the central nervous system extracellular microenvironment. Neuroscience 2012, 217, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozar, R. ADAMTS-13 in traumatic brain injury? Blood 2018, 132, 985–986. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Scully, M.; Singer, M. The role of ADAMTS-13 in the coagulopathy of sepsis. J. Thromb. Haemost. 2018, 16, 646–651. [Google Scholar] [CrossRef]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; Lopez, J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [Green Version]
- Hussein, E.; Teruya, J. Evaluating the impact of the ABO blood group on the clinical outcome of thrombotic thrombocytopenic purpura associated with severe ADAMTS13 deficiency. Vox Sang. 2017, 112, 434–442. [Google Scholar] [CrossRef]
- Schuppner, R.; Dirks, M.; Grosse, G.M.; Bockmann, M.; Goetz, F.; Pasedag, T.; Bode-Boger, S.M.; Martens-Lobenhoffer, J.; Budde, U.; Lanfermann, H.; et al. ADAMTS-13 activity predicts outcome in acute ischaemic stroke patients undergoing endovascular treatment. Thromb. Haemost. 2018, 118, 758–767. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, W.; Zhou, Y.; Hilton, T.; Zhao, Z.; Liu, W.; Wang, M.; Yeon, J.; Houck, K.; Thiagarajan, P.; et al. Von Willebrand factor enhances microvesicle-induced vascular leakage and coagulopathy in mice with traumatic brain injury. Blood 2018, 132, 1075–1084. [Google Scholar] [CrossRef]
- Cai, P.; Luo, H.; Xu, H.; Zhu, X.; Xu, W.; Dai, Y.; Xiao, J.; Cao, Y.; Zhao, Y.; Zhao, B.Q.; et al. Recombinant ADAMTS 13 attenuates brain injury after intracerebral hemorrhage. Stroke 2015, 46, 2647–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, K.; Denorme, F.; Salles, C., II; De Meyer, S.F.; Lane, D.A. Enhanced activity of an ADAMTS-13 variant (R568K/F592Y/R660K/Y661F/Y665F) against platelet agglutination in vitro and in a murine model of acute ischemic stroke. J. Thromb. Haemost. 2018, 16, 2289–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaykasli, K.O.; Oohashi, T.; Hirohata, S.; Hatipoglu, O.F.; Inagawa, K.; Demircan, K.; Ninomiya, Y. ADAMTS9 activation by interleukin 1 beta via NFATc1 in OUMS-27 chondrosarcoma cells and in human chondrocytes. Mol. Cell. Biochem. 2009, 323, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, M.J.; Cross, A.K.; Haddock, G.; Allan, S.M.; Stock, C.J.; Woodroofe, M.N.; Buttle, D.J.; Bunning, R.A. ADAMTS-9 expression is up-regulated following transient middle cerebral artery occlusion (tMCAo) in the rat. Neurosci. Lett. 2009, 452, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.B.; Domowicz, M.S. Proteoglycans in brain development and pathogenesis. FEBS Lett. 2018, 592, 3791–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abali, O.; Gokce, E.C.; Cemil, B.; Erdogan, B.; Yonezawa, T.; Demircan, K. Early induction of ADAMTS 1, -4, -5 and -9 in IL-stimulated mouse astrocytes. Turk. Neurosurg. 2014, 24, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Demircan, K.; Yonezawa, T.; Takigawa, T.; Topcu, V.; Erdogan, S.; Ucar, F.; Armutcu, F.; Yigitoglu, M.R.; Ninomiya, Y.; Hirohata, S. ADAMTS1, ADAMTS5, ADAMTS9 and aggrecanase-generated proteoglycan fragments are induced following spinal cord injury in mouse. Neurosci. Lett. 2013, 544, 25–30. [Google Scholar] [CrossRef]
- Cua, R.C.; Lau, L.W.; Keough, M.B.; Midha, R.; Apte, S.S.; Yong, V.W. Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia 2013, 61, 972–984. [Google Scholar] [CrossRef]
- Hamel, M.G.; Ajmo, J.M.; Leonardo, C.C.; Zuo, F.; Sandy, J.D.; Gottschall, P.E. Multimodal signaling by the ADAMTSs (a disintegrin and metalloproteinase with thrombospondin motifs) promotes neurite extension. Exp. Neurol. 2008, 210, 428–440. [Google Scholar] [CrossRef] [Green Version]
- Demircan, K.; Topcu, V.; Takigawa, T.; Akyol, S.; Yonezawa, T.; Ozturk, G.; Ugurcu, V.; Hasgul, R.; Yigitoglu, M.R.; Akyol, O.; et al. ADAMTS4 and ADAMTS5 knockout mice are protected from versican but not aggrecan or brevican proteolysis during spinal cord injury. BioMed Res. Int. 2014, 2014, 693746. [Google Scholar] [CrossRef]
- Pruvost, M.; Lepine, M.; Leonetti, C.; Etard, O.; Naveau, M.; Agin, V.; Docagne, F.; Maubert, E.; Ali, C.; Emery, E.; et al. ADAMTS-4 in oligodendrocytes contributes to myelination with an impact on motor function. Glia 2017, 65, 1961–1975. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.W.; Hill, R.A. Myelin plasticity in adulthood and aging. Neurosci. Lett. 2020, 715, 134645. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Richbourgh, B.; Jia, T.; Liu, C. ADAMTS-12: A multifaced metalloproteinase in arthritis and inflammation. Mediators Inflamm. 2014, 2014, 649718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhleisen, T.W.; Mattheisen, M.; Strohmaier, J.; Degenhardt, F.; Priebe, L.; Schultz, C.C.; Breuer, R.; Meier, S.; Hoffmann, P.; Investigators, G.; et al. Association between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorder. Schizophr. Res. 2012, 138, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Oruc, L.; Kapur-Pojskic, L.; Ramic, J.; Pojskic, N.; Bajrovic, K. Assessment of relatedness between neurocan gene as bipolar disorder susceptibility locus and schizophrenia. Bosn. J. Basic Med. Sci. 2012, 12, 245–248. [Google Scholar] [CrossRef]
- Schultz, C.C.; Muhleisen, T.W.; Nenadic, I.; Koch, K.; Wagner, G.; Schachtzabel, C.; Siedek, F.; Nothen, M.M.; Rietschel, M.; Deufel, T.; et al. Common variation in NCAN, a risk factor for bipolar disorder and schizophrenia, influences local cortical folding in schizophrenia. Psychol. Med. 2014, 44, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Bespalova, I.N.; Angelo, G.W.; Ritter, B.P.; Hunter, J.; Reyes-Rabanillo, M.L.; Siever, L.J.; Silverman, J.M. Genetic variations in the ADAMTS12 gene are associated with schizophrenia in Puerto Rican patients of Spanish descent. Neuromol.Med. 2012, 14, 53–64. [Google Scholar] [CrossRef]
- Koike, A.; Nishida, N.; Inoue, I.; Tsuji, S.; Tokunaga, K. Genome-wide association database developed in the Japanese Integrated Database Project. J. Hum. Genet. 2009, 54, 543–546. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.; Kubo, K.I.; Nakajima, K. Reelin and neuropsychiatric disorders. Front. Cell. Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Ogino, H.; Hisanaga, A.; Kohno, T.; Kondo, Y.; Okumura, K.; Kamei, T.; Sato, T.; Asahara, H.; Tsuiji, H.; Fukata, M.; et al. Secreted metalloproteinase ADAMTS-3 inactivates Reelin. J. Neurosci. 2017, 37, 3181–3191. [Google Scholar] [CrossRef]
- Yamakage, Y.; Tsuiji, H.; Kohno, T.; Ogino, H.; Saito, T.; Saido, T.C.; Hattori, M. Reducing ADAMTS-3 inhibits smyloid beta feposition in App knock-in mouse. Biol. Pharm. Bull. 2019, 42, 354–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakage, Y.; Kato, M.; Hongo, A.; Ogino, H.; Ishii, K.; Ishizuka, T.; Kamei, T.; Tsuiji, H.; Miyamoto, T.; Oishi, H.; et al. A disintegrin and metalloproteinase with thrombospondin motifs 2 cleaves and inactivates Reelin in the postnatal cerebral cortex and hippocampus, but not in the cerebellum. Mol. Cell. Neurosci. 2019, 100, 103401. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Sun, X.Y.; Guo, T.J.; Barot, E.; Wang, D.F.; Yan, L.L.; Ni, D.W.; Huang, N.H.; Xie, Q.; Zeng, J.; et al. Association on DISC1 SNPs with schizophrenia risk: A meta-analysis. Psychiatry Res. 2018, 270, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Jumpertz, T.; Huttenrauch, M.; Ogorek, I.; Gerber, H.; Storck, S.E.; Zampar, S.; Dimitrov, M.; Lehmann, S.; Lepka, K.; et al. The metalloprotease ADAMTS4 generates N-truncated Abeta4-x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 239–257. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| ADAMTS | Known Substrates | Neuronal Process/Disorder |
|---|---|---|
| ADAMTS-1 | Versican; brevican | Stroke [93]; spinal cord injury [117]; neuroplasticity [104]; inflammation [93,116,117]; Down’s syndrome [87]; Alzheimer’s disease [87] |
| ADAMTS-3 | Reelin | Alzheimer’s disease [130,131]; schizophrenia [130] |
| ADAMTS-4 | Versican; aggrecan; reelin; brevican | Stroke [93]; spinal cord injury [117]; neuroplasticity [83,119]; inflammation [93,116,117]; myelination [121]; Alzheimer’s disease [83,134]; schizophrenia [56] |
| ADAMTS-5 | Versican; aggrecan; reelin; brevican | Stroke [93]; spinal cord injury [117]; neuroplasticity [83]; inflammation [116,117]; Alzheimer’s disease [83] |
| ADAMTS-9 | Versican | Stroke [114]; spinal cord injury [117]; inflammation [113,116,117] |
| ADAMTS-12 | Neurocan | Inflammation [7,123]; schizophrenia [7,124,127] |
| ADAMTS-13 | von Willebrand factor | Inflammation [111]; stroke [109,110,112] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamedi, Y.; Fontanil, T.; Cobo, T.; Cal, S.; Obaya, A.J. New Insights into ADAMTS Metalloproteases in the Central Nervous System. Biomolecules 2020, 10, 403. https://doi.org/10.3390/biom10030403
Mohamedi Y, Fontanil T, Cobo T, Cal S, Obaya AJ. New Insights into ADAMTS Metalloproteases in the Central Nervous System. Biomolecules. 2020; 10(3):403. https://doi.org/10.3390/biom10030403
Chicago/Turabian StyleMohamedi, Yamina, Tania Fontanil, Teresa Cobo, Santiago Cal, and Alvaro J. Obaya. 2020. "New Insights into ADAMTS Metalloproteases in the Central Nervous System" Biomolecules 10, no. 3: 403. https://doi.org/10.3390/biom10030403
APA StyleMohamedi, Y., Fontanil, T., Cobo, T., Cal, S., & Obaya, A. J. (2020). New Insights into ADAMTS Metalloproteases in the Central Nervous System. Biomolecules, 10(3), 403. https://doi.org/10.3390/biom10030403