Epicardial TGFβ and BMP Signaling in Cardiac Regeneration: What Lesson Can We Learn from the Developing Heart?




{kind=link}
{kind=link}
{kind=link}
Abstract
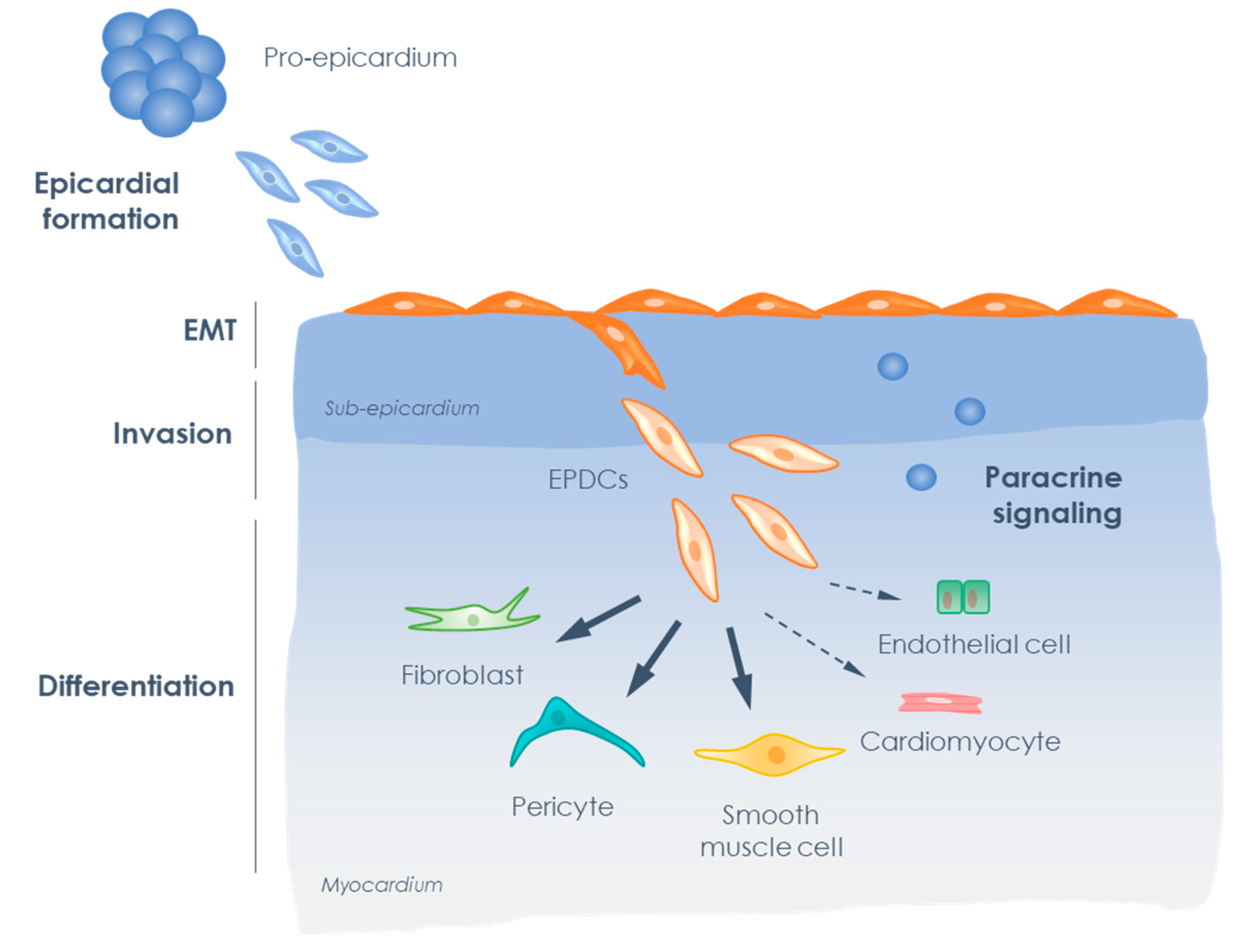
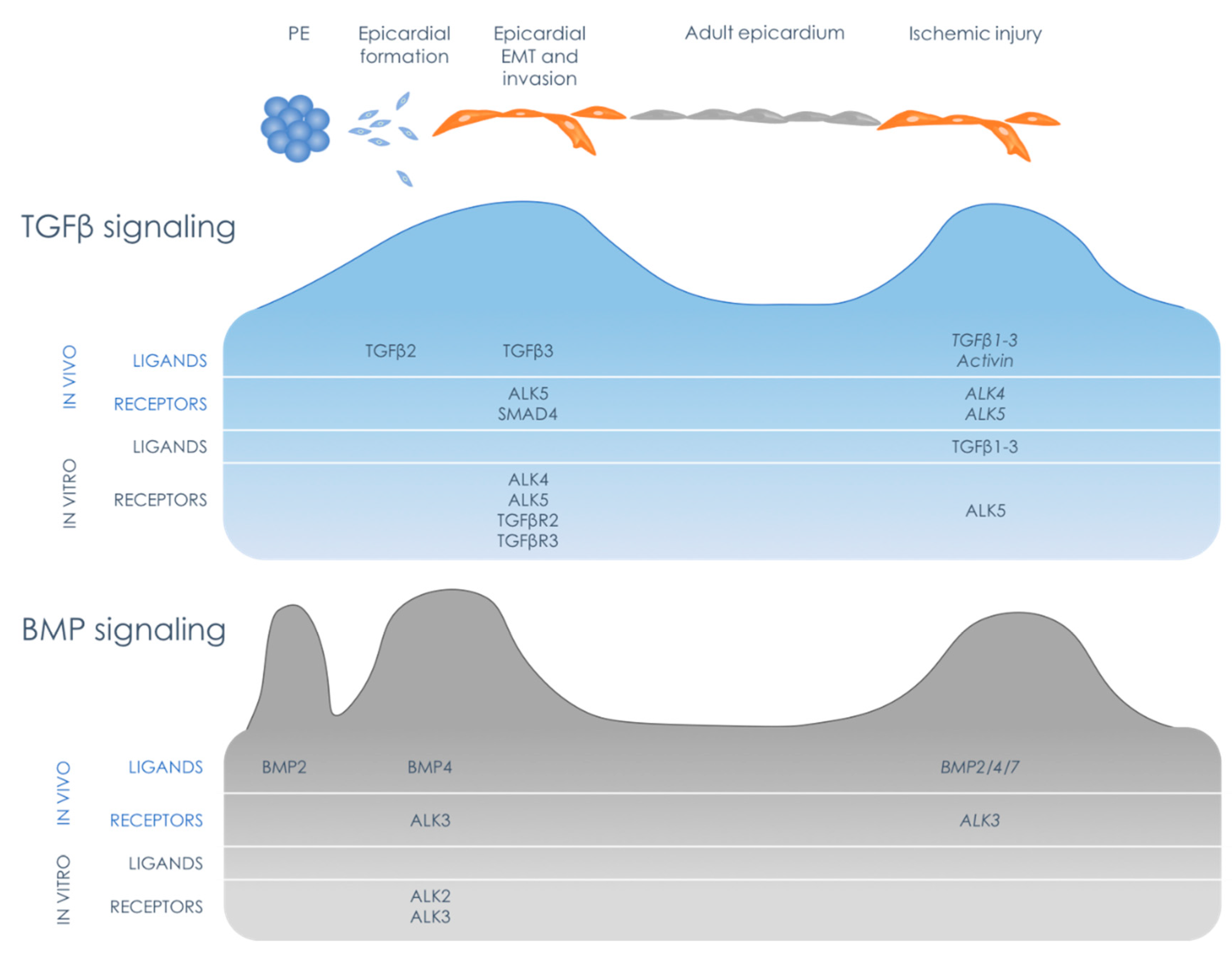
:1. Introduction: The Epicardial Timeline
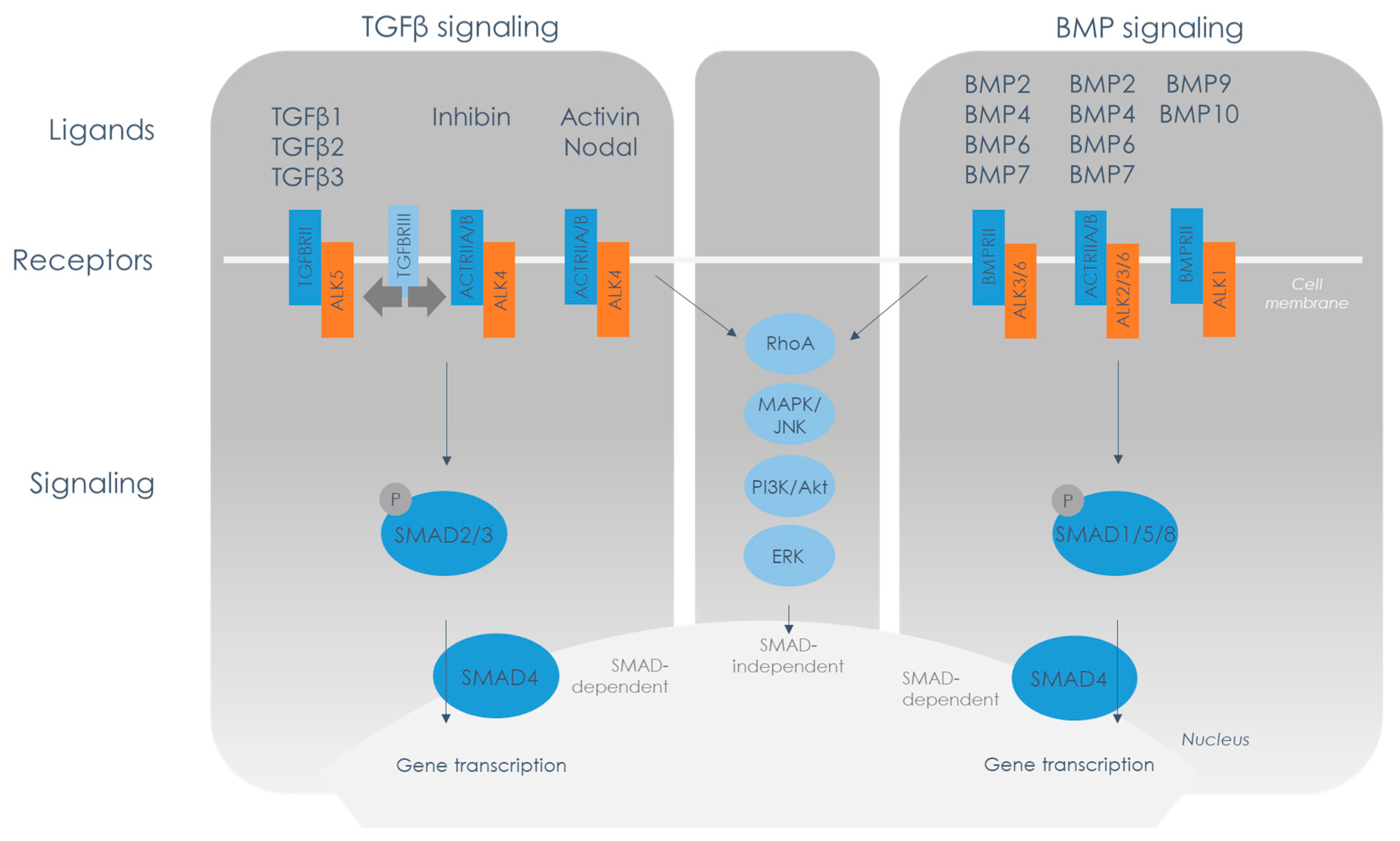
2. TGFβ Family Signaling Overview
3. Epicardium in Development and Disease
4. TGFβ Signaling in Epicardial Behavior during Cardiac Development
4.1. Expression of TGFβ Members in the Epicardium during Development
4.2. Functional Role of TGFβ in the Epicardium during Development
4.3. Downstream Signaling Mechanisms of TGFβ in the Epicardium during Development
4.4. The Role of Activin Signaling in Epicardial Behavior during Cardiac Development
5. BMP Signaling in Epicardial Behavior Cardiac Development
6. The Role of Accessory Receptors Endoglin and TGFβR3 in Developmental Epicardial Behavior
7. TGFβ Family in the Epicardial Response to Cardiac Injury
7.1. Epicardial TGFβ Signaling in the Injured Heart
7.2. Epicardial BMP Signaling in the Injured Heart
8. Epicardial TGFβ and BMP Signaling as a Target for Regenerative Therapy?
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kurkiewicz, T. O histogenezie mięśnia sercowego zwierząt kręgowych. Bull. I’ Acad. Sci. Cracovie 1909, 148–191. [Google Scholar]
- Manasek, F. Embryonic development of the heart - Formation of the Epicardium. J. Embryol. Exp. Morph. 1969, 22, 333–348. [Google Scholar] [PubMed]
- Virágh, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Káimán, F. Early development of quail heart epicardium and associated vascular and glandular structures. Anat. Embryol. (Berl). 1993, 188, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Viragh, S.; Challice, C.E. The origin of the epicardium and the embryonic myocardial circulation in the mouse. Anat. Rec. 1981, 201, 157–168. [Google Scholar] [CrossRef]
- Pérez-Pomares, J.M.; Macías, D.; García-Garrido, L.; Muñoz-Chápuli, R. Contribution of the primitive epicardium to the subepicardial mesenchyme in hamster and chick embryos. Dev. Dyn. 1997, 210, 96–105. [Google Scholar] [CrossRef]
- Mikawa, T.; Fischman, D.A. Retroviral analysis of cardiac morphogenesis: Discontinuous formation of coronary vessels. Proc. Natl. Acad. Sci. USA 1992, 89, 9504–9508. [Google Scholar] [CrossRef] [Green Version]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Dettman, R.W.; Denetclaw, W.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Poelmann, R.E. Epicardium derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 2010, 14, 1056–1060. [Google Scholar] [CrossRef]
- Kelder, T.P.; Duim, S.N.; Vicente-Steijn, R.; Végh, A.M.D.; Kruithof, B.P.T.; Smits, A.M.; van Bavel, T.C.; Bax, N.A.M.; Schalij, M.J.; Gittenberger-de Groot, A.C.; et al. The epicardium as modulator of the cardiac autonomic response during early development. J. Mol. Cell. Cardiol. 2015, 89, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.H.P.; Chang, T.-C.; Kang, J.-O.; Choudhary, B.; Makita, T.; Tran, C.M.; Burch, J.B.E.; Eid, H.; Sucov, H.M. Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev. Biol. 2002, 250, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Weeke-Klimp, A.; Bax, N.A.M.; Bellu, A.R.; Winter, E.M.; Vrolijk, J.; Plantinga, J.; Maas, S.; Brinker, M.; Mahtab, E.A.F.; Gittenberger-de Groot, A.C.; et al. Epicardium-derived cells enhance proliferation, cellular maturation and alignment of cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 49, 606–616. [Google Scholar] [CrossRef]
- Männer, J.; Schlueter, J.; Brand, T. Experimental analyses of the function of the proepicardium using a new microsurgical procedure to induce loss-of-proepicardial-function in chick embryos. Dev. Dyn. 2005, 233, 1454–1463. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.-P.F.M.; Bergwerff, M.; Mentink, M.M.T.; Poelmann, R.E. Epicardial Outgrowth Inhibition Leads to Compensatory Mesothelial Outflow Tract Collar and Abnormal Cardiac Septation and Coronary Formation. Circ. Res. 2000, 87, 969–971. [Google Scholar] [CrossRef] [Green Version]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A Dynamic Epicardial Injury Response Supports Progenitor Cell Activity during Zebrafish Heart Regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef]
- van Tuyn, J.; Atsma, D.E.; Winter, E.M.; van der Velde-van Dijke, I.; Pijnappels, D.A.; Bax, N.A.; Knaän-Shanzer, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; van der Laarse, A.; et al. Epicardial Cells of Human Adults Can Undergo an Epithelial-to-Mesenchymal Transition and Obtain Characteristics of Smooth Muscle Cells In Vitro. Stem Cells 2007, 25, 271–278. [Google Scholar] [CrossRef]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.-H.; Butterfield, C.; Lin, R.-Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Investig. 2011, 121, 1894–1904. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.; Riley, P. Epicardium-Derived Heart Repair. J. Dev. Biol. 2014, 2, 84–100. [Google Scholar] [CrossRef]
- Witman, N.; Murtuza, B.; Davis, B.; Arner, A.; Morrison, J.I. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev. Biol. 2011, 354, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Wu, Q.; Zhang, Y.; Wiens, K.M.; Huang, Y.; Rubin, N.; Shimada, H.; Handin, R.I.; Chao, M.Y.; Tuan, T.-L.; et al. PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc. Natl. Acad. Sci. USA 2010, 107, 17206–17210. [Google Scholar] [CrossRef] [Green Version]
- Teotia, P.; Van Hook, M.J.; Fischer, D.; Ahmad, I. Human retinal ganglion cell axon regeneration by recapitulating developmental mechanisms: Effects of recruitment of the mTOR pathway. Development 2019, 146, dev178012. [Google Scholar] [CrossRef] [Green Version]
- McDermott, A.M.; Herberg, S.; Mason, D.E.; Collins, J.M.; Pearson, H.B.; Dawahare, J.H.; Tang, R.; Patwa, A.N.; Grinstaff, M.W.; Kelly, D.J.; et al. Recapitulating bone development through engineered mesenchymal condensations and mechanical cues for tissue regeneration. Sci. Transl. Med. 2019, 11, eaav7756. [Google Scholar] [CrossRef]
- Kahata, K.; Dadras, M.S.; Moustakas, A. TGF-β Family Signaling in Epithelial Differentiation and Epithelial–Mesenchymal Transition. Cold Spring Harb. Perspect. Biol. 2018, 10, a022194. [Google Scholar] [CrossRef] [Green Version]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-β in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar]
- Wu, M.Y.; Hill, C.S. TGF-β Superfamily Signaling in Embryonic Development and Homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Gentry, L.E.; Webb, N.R.; Lim, G.J.; Brunner, A.M.; Ranchalis, J.E.; Twardzik, D.R.; Lioubin, M.N.; Marquardt, H.; Purchio, A.F. Type 1 transforming growth factor beta: Amplified expression and secretion of mature and precursor polypeptides in Chinese hamster ovary cells. Mol. Cell. Biol. 1987, 7, 3418–3427. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.M.; Laprise, M.H.; Blanchette, F.; Gentry, L.E.; Leduc, R. Processing of transforming growth factor β1 precursor by human furin convertase. J. Biol. Chem. 1995, 270, 10618–10624. [Google Scholar] [CrossRef] [Green Version]
- Sengle, G.; Ono, R.N.; Lyons, K.M.; Bächinger, H.P.; Sakai, L.Y. A New Model for Growth Factor Activation: Type II Receptors Compete with the Prodomain for BMP-7. J. Mol. Biol. 2008, 381, 1025–1039. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.A.; Zhao, Q.; Baker, K.A.; Naik, C.; Chen, C.; Pukac, L.; Singh, M.; Tsareva, T.; Parice, Y.; Mahoney, A.; et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005, 280, 25111–25118. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Salmon, R.M.; Upton, P.D.; Wei, Z.; Lawera, A.; Davenport, A.P.; Morrell, N.W.; Li, W. The prodomain-bound form of bone morphogenetic protein 10 is biologically active on endothelial cells. J. Biol. Chem. 2016, 291, 2954–2966. [Google Scholar] [CrossRef] [Green Version]
- Sengle, G.; Ono, R.N.; Sasaki, T.; Sakai, L.Y. Prodomains of transforming growth factor β (TGFβ) Superfamily members specify different functions: Extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 2011, 286, 5087–5099. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; ten Dijke, P. TGF-β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2017, 10, 1–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Chen, Y.G. TGF-β signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-J.; Klausen, C.; Li, Y.; Zhu, H.; Wang, Y.-L.; Leung, P.C.K. Bone morphogenetic protein 2 promotes human trophoblast cell invasion by upregulating N-cadherin via non-canonical SMAD2/3 signaling. Cell Death Dis. 2018, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P.; Ananth, S.; Knebelmann, B.; Gruning, W.; Dhanabal, M.; et al. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin Receptor-like Kinase (ALK)1 Is an Antagonistic Mediator of Lateral TGFβ/ALK5 Signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Männer, J. Experimental study on the formation of the epicardium in chick embryos. Anat. Embryol. (Berl). 1993, 187, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Velecela, V.; Torres-Cano, A.; García-Melero, A.; Ramiro-Pareta, M.; Müller-Sánchez, C.; Segarra-Mondejar, M.; Chau, Y.; Campos-Bonilla, B.; Reina, M.; Soriano, F.X.; et al. Epicardial cell shape and maturation are regulated by Wt1 via transcriptional control of Bmp4. Development 2019, 146, dev178723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Smith, C.L.; Hall, J.A.; Lee, I.; Luby-Phelps, K.; Tallquist, M.D. Epicardial Spindle Orientation Controls Cell Entry into the Myocardium. Dev. Cell 2010, 19, 114–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Pomares, J.M.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; González-Iriarte, M.; Muñoz-Chápuli, R.; Wessels, A.; Muoz-Chápuli, R.; Wessels, A.; Muñoz-Chápuli, R.; et al. Experimental Studies on the Spatiotemporal Expression of WT1 and RALDH2 in the Embryonic Avian Heart: A Model for the Regulation of Myocardial and Valvuloseptal Development by Epicardially Derived Cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef] [Green Version]
- Von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef]
- von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. Wt1 regulates epicardial epithelial to mesenchymal transition through beta-catenin and retinoic acid signaling pathways. Dev. Biol. 2011, 356, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.M.; Dronkers, E.; Goumans, M.-J.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef]
- Vega-Hernández, M.; Kovacs, A.; de Langhe, S.; Ornitz, D.M. FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development 2011, 138, 3331–3340. [Google Scholar] [CrossRef] [Green Version]
- Zamora, M.; Männer, J.; Ruiz-Lozano, P. Epicardium-derived progenitor cells require β-catenin for coronary artery formation. Proc. Natl. Acad. Sci. USA 2007, 104, 18109–18114. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; von Gise, A.; Ma, Q.; Hu, Y.W.; Pu, W.T. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Biol. 2010, 338, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Wessels, A.; van den Hoff, M.J.B.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, M.M.; Phelps, A.L.; van den Hoff, M.J.B.; Wessels, A. The epicardium and the development of the atrioventricular junction in the murine heart. J. Dev. Biol. 2014, 2, 1–17. [Google Scholar] [CrossRef]
- Merki, E.; Zamora, M.; Raya, A.; Kawakami, Y.; Wang, J.; Zhang, X.; Burch, J.; Kubalak, S.W.; Kaliman, P.; Belmonte, J.C.I.; et al. Epicardial retinoid X receptor is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 2005, 102, 18455–18460. [Google Scholar] [CrossRef] [Green Version]
- Lavine, K.J.; Yu, K.; White, A.C.; Zhang, X.; Smith, C.; Partanen, J.; Ornitz, D.M. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 2005, 8, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Kolander, K.D.; Holtz, M.L.; Cossette, S.M.; Duncan, S.A.; Misra, R.P. Epicardial GATA factors regulate early coronary vascular plexus formation. Dev. Biol. 2014, 386, 204–215. [Google Scholar] [CrossRef] [Green Version]
- van Wijk, B.; Gunst, Q.D.; Moorman, A.F.M.; van den Hoff, M.J.B. Cardiac regeneration from activated epicardium. PLoS ONE 2012, 7, e44692. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Molin, D.G.M.; Bartram, U.; Van der Heiden, K.; Van Iperen, L.; Speer, C.P.; Hierck, B.P.; Poelmann, R.E.; Gittenberger-de-Groot, A.C. Expression patterns of Tgfβ1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev. Dyn. 2003, 227, 431–444. [Google Scholar] [CrossRef]
- Compton, L.A.; Potash, D.A.; Mundell, N.A.; Barnett, J.V. Transforming growth factor-β induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Dev. Dyn. 2006, 235, 82–93. [Google Scholar] [CrossRef]
- Engelmann, G.L. Coordinate gene expression during neonatal rat heart development. A possible role for the myocyte in extracellular matrix biogenesis and capillary angiogenesis. Cardiovasc. Res. 1993, 27, 1598–1605. [Google Scholar] [CrossRef]
- Hill, C.R.; Sanchez, N.S.; Love, J.D.; Arrieta, J.A.; Hong, C.C.; Brown, C.B.; Austin, A.F.; Barnett, J.V. BMP2 signals loss of epithelial character in epicardial cells but requires the Type III TGFβ receptor to promote invasion. Cell. Signal. 2012, 24, 1012–1022. [Google Scholar] [CrossRef] [Green Version]
- Sridurongrit, S.; Larsson, J.; Schwartz, R.; Ruiz-Lozano, P.; Kaartinen, V. Signaling via the Tgf-β type I receptor Alk5 in heart development. Dev. Biol. 2008, 322, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Lüdtke, T.H.; Rudat, C.; Kurz, J.; Häfner, R.; Greulich, F.; Wojahn, I.; Aydoğdu, N.; Mamo, T.M.; Kleppa, M.-J.; Trowe, M.-O.; et al. Mesothelial mobilization in the developing lung and heart differs in timing, quantity, and pathway dependency. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L767–L783. [Google Scholar] [CrossRef]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar]
- Kruithof, B.P.T.; Kruithof-De-Julio, M.; Poelmann, R.E.; Gittenberger-De-Groot, A.C.; Gaussin, V.; Goumans, M.J. Remodeling of the myocardium in early trabeculation and cardiac valve formation; a role for TGFβ2. Int. J. Dev. Biol. 2013, 57, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF–β3 indicates defects of epithelial–mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef]
- Azhar, M.; Schultz, J.E.J.; Grupp, I.; Dorn, G.W.; Meneton, P.; Molin, D.G.M.; Gittenberger-de Groot, A.C.; Doetschman, T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003, 14, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef]
- Larsson, J.; Goumans, M.J.; Sjöstrand, L.J.; Van Rooijen, M.A.; Ward, D.; Levéen, P.; Xu, X.; Ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef] [Green Version]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Letterio, J.; Geiser, A.; Kulkarni, A.; Roche, N.; Sporn, M.; Roberts, A. Maternal rescue of transforming growth factor-beta 1 null mice. Science 1994, 264, 1936–1938. [Google Scholar] [CrossRef]
- Daniel, C.W.; Silberstein, G.B.; Van Horn, K.; Strickland, P.; Robinson, S. TGF-β1-induced inhibition of mouse mammary ductal growth: Developmental specificity and characterization. Dev. Biol. 1989, 135, 20–30. [Google Scholar] [CrossRef]
- Migdalska, A.; Molineux, G.; Demuynck, H.; Evans, G.S.; Ruscettp, F.; Dexter, T.M. Growth inhibitory effects of transforming growth factor-β1 in vivo. Growth Factors 1991, 4, 239–245. [Google Scholar] [CrossRef]
- MacNeill, C.; French, R.; Evans, T.; Wessels, A.; Burch, J.B.E. Modular regulation of cGATA-5 gene expression in the developing heart and gut. Dev. Biol. 2000, 217, 62–76. [Google Scholar] [CrossRef]
- Olivey, H.E.; Mundell, N.A.; Austin, A.F.; Barnett, J.V. Transforming growth factor-β stimulates epithelial–mesenchymal transformation in the proepicardium. Dev. Dyn. 2006, 235, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Austin, A.F.; Compton, L.A.; Love, J.D.; Brown, C.B.; Barnett, J.V. Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFβ. Dev. Dyn. 2008, 237, 366–376. [Google Scholar] [CrossRef]
- Dronkers, E.; Moerkamp, A.T.; van Herwaarden, T.; Goumans, M.-J.; Smits, A.M. The Isolation and Culture of Primary Epicardial Cells Derived from Human Adult and Fetal Heart Specimens. J. Vis. Exp. 2018, 134, e57370. [Google Scholar] [CrossRef]
- Moerkamp, A.T.; Lodder, K.; van Herwaarden, T.; Dronkers, E.; Dingenouts, C.K.E.; Tengström, F.C.; van Brakel, T.J.; Goumans, M.-J.; Smits, A.M. Human fetal and adult epicardial-derived cells: A novel model to study their activation. Stem Cell Res. Ther. 2016, 7, 174. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef]
- Dokic, D.; Dettman, R.W. VCAM-1 inhibits TGFβ stimulated epithelial-mesenchymal transformation by modulating Rho activity and stabilizing intercellular adhesion in epicardial mesothelial cells. Dev. Biol. 2006, 299, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Morabito, C.J.; Dettman, R.W.; Kattan, J.; Collier, J.M.; Bristow, J. Positive and Negative Regulation of Epicardial–Mesenchymal Transformation during Avian Heart Development. Dev. Biol. 2001, 234, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Aakre, M.E.; Edgerton, M.; Shyr, Y.; Bhowmick, N.A.; Moses, H.L. Transforming growth factor beta-regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res. 2003, 5, R187–R198. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Barnett, J.; Watanabe, M.; Ramírez-Bergeron, D. Hypoxia Supports Epicardial Cell Differentiation in Vascular Smooth Muscle Cells through the Activation of the TGFβ Pathway. J. Cardiovasc. Dev. Dis. 2018, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, N.S.; Barnett, J.V. TGFβ and BMP-2 regulate epicardial cell invasion via TGFβR3 activation of the Par6/Smurf1/RhoA pathway. Cell. Signal. 2012, 24, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Narumiya, S.; Thumkeo, D. Rho signaling research: History, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B.; Vogler, R.; Zimmermann, K.; Fujii, M.; Anzano, M.B.; Schäfer-Korting, M.; Roberts, A.B.; Kleuser, B. Lysophosphatidic acid interacts with transforming growth factor-β signaling to mediate keratinocyte growth arrest and chemotaxis. J. Investig. Dermatol. 2004, 123, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Trembley, M.A.; Velasquez, L.S.; de Mesy Bentley, K.L.; Small, E.M. Myocardin-related transcription factors control the motility of epicardium-derived cells and the maturation of coronary vessels. Development 2015, 142, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Craig, E.A.; Austin, A.F.; Vaillancourt, R.R.; Barnett, J.V.; Camenisch, T.D. TGFβ2-mediated production of hyaluronan is important for the induction of epicardial cell differentiation and invasion. Exp. Cell Res. 2010, 316, 3397–3405. [Google Scholar] [CrossRef] [Green Version]
- Namwanje, M.; Brown, C.W. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a021881. [Google Scholar] [CrossRef]
- Oh, S.P.; Li, E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 1997, 11, 1812–1826. [Google Scholar] [CrossRef] [Green Version]
- Roessler, E.; Pei, W.; Ouspenskaia, M.V.; Karkera, J.D.; Veléz, J.I.; Banerjee-Basu, S.; Gibney, G.; Lupo, P.J.; Mitchell, L.E.; Towbin, J.A.; et al. Cumulative ligand activity of NODAL mutations and modifiers are linked to human heart defects and holoprosencephaly. Mol. Genet. Metab. 2009, 98, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Davis, D.A.; Burdette, J.E. Activin A stimulates migration of the fallopian tube epithelium, an origin of high-grade serous ovarian cancer, through non-canonical signaling. Cancer Lett. 2017, 391, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.; Ozden, O.; Akagi, N.; Carroll, T.; Principe, D.R.; Staudacher, J.J.; Spehlmann, M.E.; Eckmann, L.; Grippo, P.J.; Jung, B. Activin and TGFβ use diverging mitogenic signaling in advanced colon cancer. Mol. Cancer 2015, 14, 182. [Google Scholar] [CrossRef] [Green Version]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Suzuki, M.; Nishino, Y.; Funaba, M. Regulatory expression of genes related to metastasis by TGF-β and activin A in B16 murine melanoma cells. Mol. Biol. Rep. 2010, 37, 1279–1286. [Google Scholar] [CrossRef]
- Basu, M.; Bhattacharya, R.; Ray, U.; Mukhopadhyay, S.; Chatterjee, U.; Roy, S.S. Invasion of ovarian cancer cells is induced byPITX2-mediated activation of TGF-β and Activin-A. Mol. Cancer 2015, 14, 162. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.S.; Mjaatvedt, C.H.; Gearhart, J.D. Expression and function of activin beta A during mouse cardiac cushion tissue formation. Dev. Dyn. 1998, 212, 548–562. [Google Scholar] [CrossRef]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Kruithof, B.P.T.; van Wijk, B.; Somi, S.; Kruithof-de Julio, M.; Pérez Pomares, J.M.; Weesie, F.; Wessels, A.; Moorman, A.F.M.; van den Hoff, M.J.B. BMP and FGF regulate the differentiation of multipotential pericardial mesoderm into the myocardial or epicardial lineage. Dev. Biol. 2006, 295, 507–522. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Feliciano, J.; Tabin, C.J. Bmp2 instructs cardiac progenitors to form the heart-valve-inducing field. Dev. Biol. 2006, 295, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Gaussin, V.; Van De Putte, T.; Mishina, Y.; Hanks, M.C.; Zwijsen, A.; Huylebroeck, D.; Behringer, R.R.; Schneider, M.D. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc. Natl. Acad. Sci. USA 2002, 99, 2878–2883. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Fässler, R.; Mishina, Y.; Jiao, K.; Baldwin, H.S. Essential functions of Alk3 during AV cushion morphogenesis in mouse embryonic hearts. Dev. Biol. 2007, 301, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, M.M.; Boukens, B.J.D.; Phelps, A.L.; Brown, C.-L.M.; Toomer, K.A.; Burns, T.A.; Mukherjee, R.D.; Norris, R.A.; Trusk, T.C.; van den Hoff, M.J.B.; et al. Alk3 mediated Bmp signaling controls the contribution of epicardially derived cells to the tissues of the atrioventricular junction. Dev. Biol. 2014, 396, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Ten Dijke, P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar] [CrossRef]
- Ollauri-Ibáñez, C.; López-Novoa, J.M.; Pericacho, M. Endoglin-based biological therapy in the treatment of angiogenesis-dependent pathologies. Expert Opin. Biol. Ther. 2017, 17, 1053–1063. [Google Scholar] [CrossRef]
- Mercado-Pimentel, M.E.; Hubbard, A.D.; Runyan, R.B. Endoglin and Alk5 regulate epithelial-mesenchymal transformation during cardiac valve formation. Dev. Biol. 2007, 304, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Valeria, B.; Maddalena, G.; Enrica, V.; Onofrio, T.; Gaetano, B. Endoglin (CD105) Expression in the Human Heart Throughout Gestation: An Immunohistochemical Study. Reprod. Sci. 2008, 15, 1018–1026. [Google Scholar] [CrossRef]
- Hu, J.; Guan, W.; Yan, L.; Ye, Z.; Wu, L.; Xu, H. Cancer Stem Cell Marker Endoglin (CD105) Induces Epithelial Mesenchymal Transition (EMT) but Not Metastasis in Clear Cell Renal Cell Carcinoma. Stem Cells Int. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Bax, N.A.M.; Oorschot, A.A.M.; Maas, S.; Braun, J.; Tuyn, J.; Vries, A.A.F.; Groot, A.C.G.; Goumans, M.-J. In vitro epithelial-to-mesenchymal transformation in human adult epicardial cells is regulated by TGFβ-signaling and WT1. Basic Res. Cardiol. 2011, 106, 829–847. [Google Scholar] [CrossRef] [Green Version]
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan presents ligand to the TGFβ signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Kirkbride, K.C.; Townsend, T.A.; Bruinsma, M.W.; Barnett, J.V.; Blobe, G.C. Bone Morphogenetic Proteins Signal through the Transforming Growth Factor-β Type III Receptor. J. Biol. Chem. 2008, 283, 7628–7637. [Google Scholar] [CrossRef] [Green Version]
- Wiater, E.; Harrison, C.A.; Lewis, K.A.; Gray, P.C.; Vale, W.W. Identification of distinct inhibin and transforming growth factor β-binding sites on betaglycan: Functional separation of betaglycan co-receptor actions. J. Biol. Chem. 2006, 281, 17011–17022. [Google Scholar] [CrossRef] [Green Version]
- Sankar, S.; Mahooti-Brooks, N.; Centrella, M.; McCarthy, T.L.; Madri, J.A. Expression of Transforming Growth Factor Type III Receptor in Vascular Endothelial Cells Increases Their Responsiveness to Transforming Growth Factor β2. J. Biol. Chem. 1995, 270, 13567–13572. [Google Scholar] [CrossRef] [Green Version]
- Cheifetz, S.; Hernandez, H.; Laiho, M.; ten Dijke, P.; Iwata, K.K.; Massagué, J. Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J. Biol. Chem. 1990, 265, 20533–20538. [Google Scholar]
- Blobe, G.C.; Schiemann, W.P.; Pepin, M.C.; Beauchemin, M.; Moustakas, A.; Lodish, H.F.; O’Connor-McCourt, M.D. Functional Roles for the Cytoplasmic Domain of the Type III Transforming Growth Factor β Receptor in Regulating Transforming Growth Factor β Signaling. J. Biol. Chem. 2001, 276, 24627–24637. [Google Scholar] [CrossRef] [Green Version]
- Compton, L.A.; Potash, D.A.; Brown, C.B.; Barnett, J.V. Coronary Vessel Development Is Dependent on the Type III Transforming Growth Factor β Receptor. Circ. Res. 2007, 101, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, N.S.; Hill, C.R.; Love, J.D.; Soslow, J.H.; Craig, E.; Austin, A.F.; Brown, C.B.; Czirok, A.; Camenisch, T.D.; Barnett, J.V. The cytoplasmic domain of TGFβR3 through its interaction with the scaffolding protein, GIPC, directs epicardial cell behavior. Dev. Biol. 2011, 358, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Allison, P.; Espiritu, D.; Camenisch, T.D. BMP2 rescues deficient cell migration in Tgfbr3(-/-) epicardial cells and requires Src kinase. Cell Adh. Migr. 2016, 10, 259–268. [Google Scholar] [CrossRef] [Green Version]
- DeLaughter, D.M.; Clark, C.R.; Christodoulou, D.C.; Seidman, C.E.; Baldwin, H.S.; Seidman, J.G.; Barnett, J.V. Transcriptional Profiling of Cultured, Embryonic Epicardial Cells Identifies Novel Genes and Signaling Pathways Regulated by TGFβR3 In Vitro. PLoS ONE 2016, 11, e0159710. [Google Scholar] [CrossRef]
- Clark, C.R.; Robinson, J.Y.; Sanchez, N.S.; Townsend, T.A.; Arrieta, J.A.; Merryman, W.D.; Trykall, D.Z.; Olivey, H.E.; Hong, C.C.; Barnett, J.V. Common pathways regulate Type III TGFβ receptor-dependent cell invasion in epicardial and endocardial cells. Cell. Signal. 2016, 28, 688–698. [Google Scholar] [CrossRef] [Green Version]
- You, H.J.; How, T.; Blobe, G.C. The type III transforming growth factor-β receptor negatively regulates nuclear factor kappa B signaling through its interaction with β-arrestin2. Carcinogenesis 2009, 30, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K.; Ehata, S.; Koinuma, D. Tumor-promoting functions of transforming growth factor-β in progression of cancer. Ups. J. Med. Sci. 2012, 117, 143–152. [Google Scholar] [CrossRef]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Frangogiannis, N. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Chuva De Sousa Lopes, S.M.; Feijen, A.; Korving, J.; Korchynskyi, O.; Larsson, J.; Karlsson, S.; Ten Dijke, P.; Lyons, K.M.; Goldschmeding, R.; Doevendans, P.; et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev. Dyn. 2004, 231, 542–550. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J. Thorac. Dis. 2017, 9, S52–S63. [Google Scholar] [CrossRef] [Green Version]
- Euler, G. Good and bad sides of TGFβ-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on TGF signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Karra, R.; Dickson, A.L.; Poss, K.D. Fibronectin is deposited by injury-activated epicardial cells and is necessary for zebrafish heart regeneration. Dev. Biol. 2013, 382, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Marro, J.; Pfefferli, C.; de Preux Charles, A.-S.; Bise, T.; Jaźwińska, A. Collagen XII Contributes to Epicardial and Connective Tissues in the Zebrafish Heart during Ontogenesis and Regeneration. PLoS ONE 2016, 11, e0165497. [Google Scholar] [CrossRef] [Green Version]
- Dogra, D.; Ahuja, S.; Kim, H.-T.; Rasouli, S.J.; Stainier, D.Y.R.; Reischauer, S. Opposite effects of Activin type 2 receptor ligands on cardiomyocyte proliferation during development and repair. Nat. Commun. 2017, 8, 1902. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.-J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C.; Aldred, M.; Brannon, C.A.; et al. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Urness, L.D.; Sorensen, L.K.; Li, D.Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 2000, 26, 328–331. [Google Scholar] [CrossRef]
- Stahls, P.F.; Lightell, D.J.; Moss, S.C.; Goldman, C.K.; Woods, T.C. Elevated serum bone morphogenetic protein 4 in patients with chronic kidney disease and coronary artery disease. J. Cardiovasc. Transl. Res. 2013, 6, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 1–15. [Google Scholar] [CrossRef]
- Wu, C.C.; Kruse, F.; Vasudevarao, M.D.; Junker, J.P.; Zebrowski, D.C.; Fischer, K.; Noël, E.S.; Grün, D.; Berezikov, E.; Engel, F.B.; et al. Spatially Resolved Genome-wide Transcriptional Profiling Identifies BMP Signaling as Essential Regulator of Zebrafish Cardiomyocyte Regeneration. Dev. Cell 2016, 36, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Navis, A.; Cox, B.D.; Dickson, A.L.; Gemberling, M.; Karra, R.; Bagnat, M.; Poss, K.D. Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 2016, 143, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Rossdeutsch, A.; Smart, N.; Dubé, K.N.; Turner, M.; Riley, P.R. Essential Role for Thymosin β4 in Regulating Vascular Smooth Muscle Cell Development and Vessel Wall Stability. Circ. Res. 2012, 111, e89–e102. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, I.; Zhang, J.; Moore-Morris, T.; Lange, S.; Shen, T.; Dalton, N.D.; Gu, Y.; Peterson, K.L.; Evans, S.M.; Chen, J. Thymosin beta 4 is dispensable for murine cardiac development and function. Circ. Res. 2012, 110, 456–464. [Google Scholar] [CrossRef]
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β-4 is essential for coronary vessel development and promotes neovascularization via adult epicardium. Ann. N. Y. Acad. Sci. 2007, 1112, 171–188. [Google Scholar] [CrossRef]
- Tandon, P.; Miteva, Y.V.; Kuchenbrod, L.M.; Cristea, I.M.; Conlon, F.L. Tcf21 regulates the specification and maturation of proepicardial cells. Development 2012, 140, 2409–2421. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.; Riley, P.R. The epicardium as a candidate for heart regeneration. Future Cardiol. 2012, 8, 53–69. [Google Scholar] [CrossRef] [Green Version]
- Quijada, P.; Trembley, M.A.; Small, E.M. The Role of the Epicardium During Heart Development and Repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dronkers, E.; Wauters, M.M.M.; Goumans, M.J.; Smits, A.M. Epicardial TGFβ and BMP Signaling in Cardiac Regeneration: What Lesson Can We Learn from the Developing Heart? Biomolecules 2020, 10, 404. https://doi.org/10.3390/biom10030404
Dronkers E, Wauters MMM, Goumans MJ, Smits AM. Epicardial TGFβ and BMP Signaling in Cardiac Regeneration: What Lesson Can We Learn from the Developing Heart? Biomolecules. 2020; 10(3):404. https://doi.org/10.3390/biom10030404
Chicago/Turabian StyleDronkers, Esther, Manon M. M. Wauters, Marie José Goumans, and Anke M. Smits. 2020. "Epicardial TGFβ and BMP Signaling in Cardiac Regeneration: What Lesson Can We Learn from the Developing Heart?" Biomolecules 10, no. 3: 404. https://doi.org/10.3390/biom10030404
APA StyleDronkers, E., Wauters, M. M. M., Goumans, M. J., & Smits, A. M. (2020). Epicardial TGFβ and BMP Signaling in Cardiac Regeneration: What Lesson Can We Learn from the Developing Heart? Biomolecules, 10(3), 404. https://doi.org/10.3390/biom10030404