Codon Harmonization of a Kir3.1-KirBac1.3 Chimera for Structural Study Optimization

Abstract
: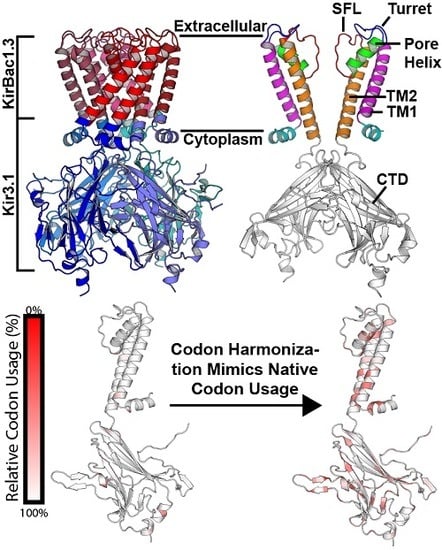
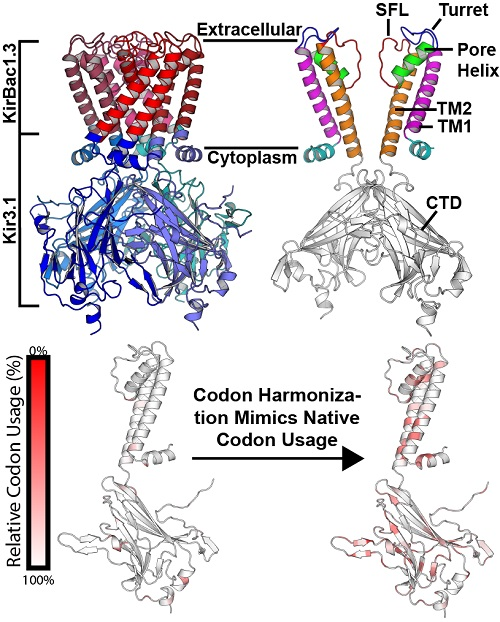{kind=link}
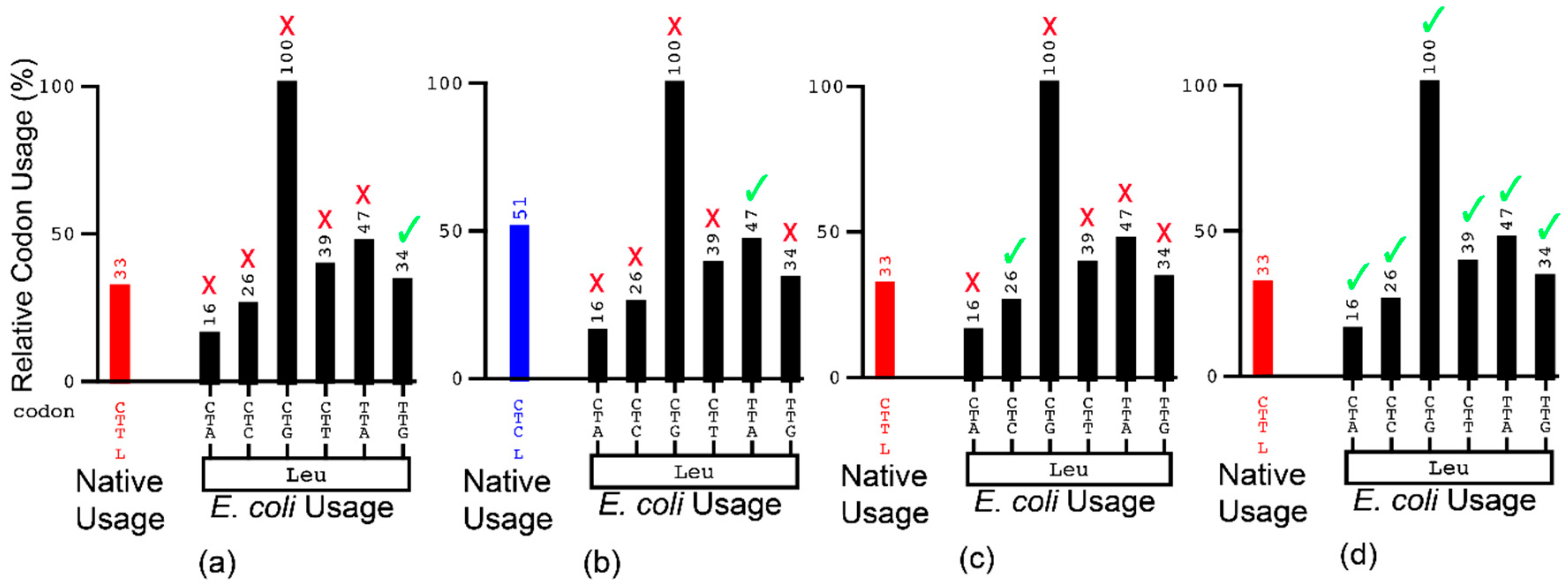{kind=link}
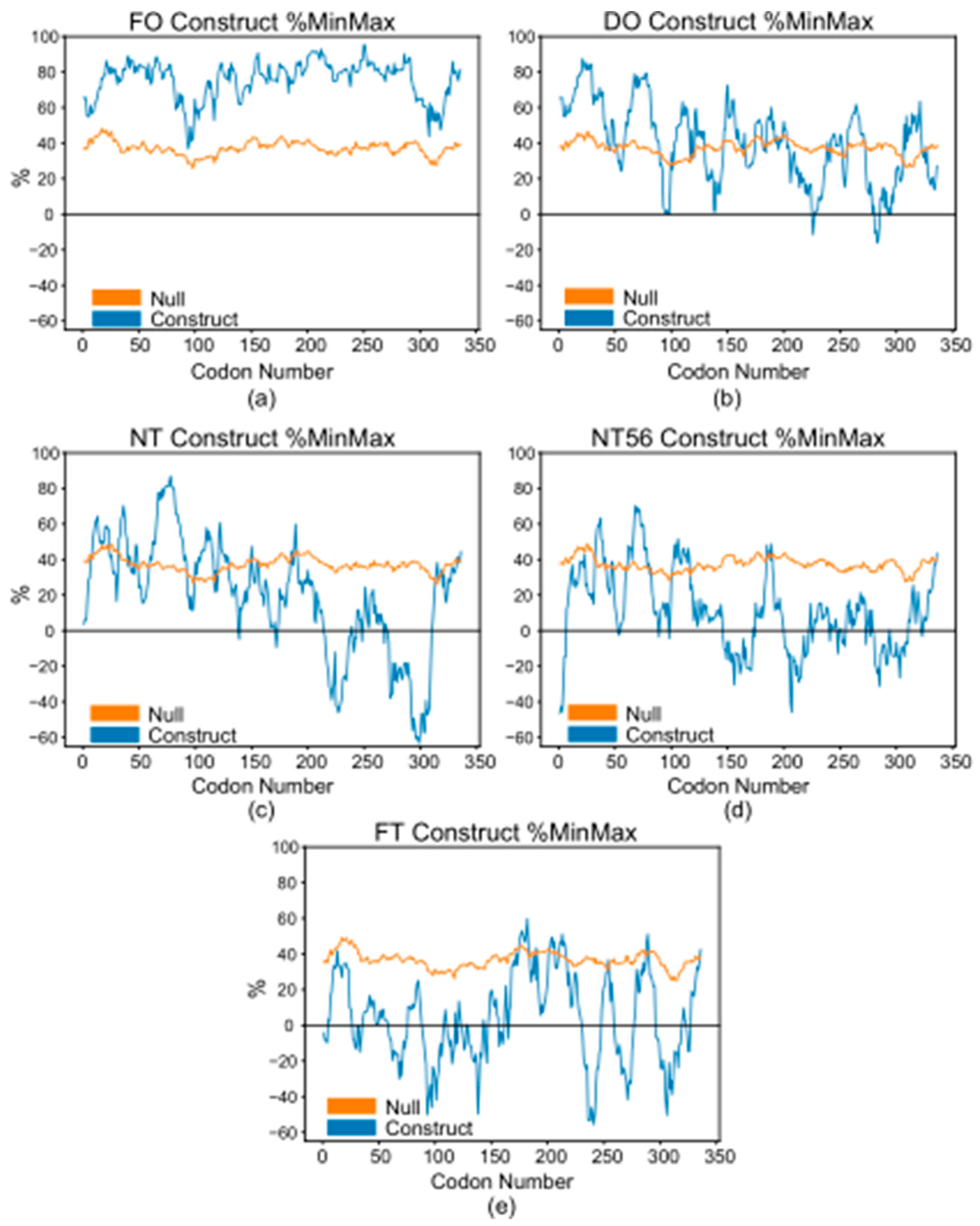{kind=link}
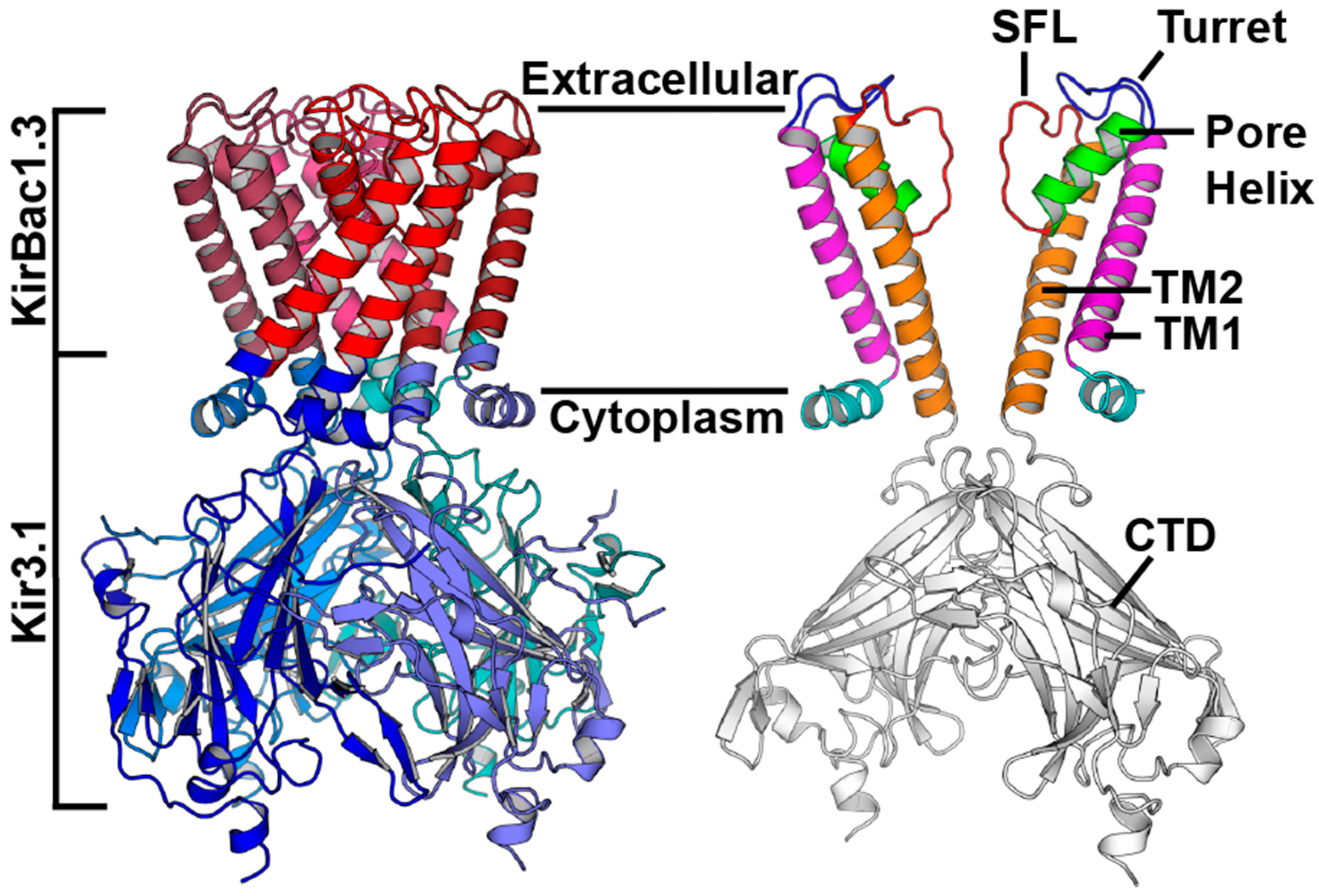{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Codon Harmonization
2.1.1. FO and DO Sequence Design
2.1.2. CW Sequence Design
2.2. Plasmid Construction
2.3. Sequence Analysis and Estimation
2.4. Expression and Purification of the GIRK Chimera
2.5. Reconstitution
2.6. Efflux Assay
2.7. NMR Measurements
2.8. Molecular Dynamics, Chemical Shift Prediction, and Torsion Angle Prediction
3. Results
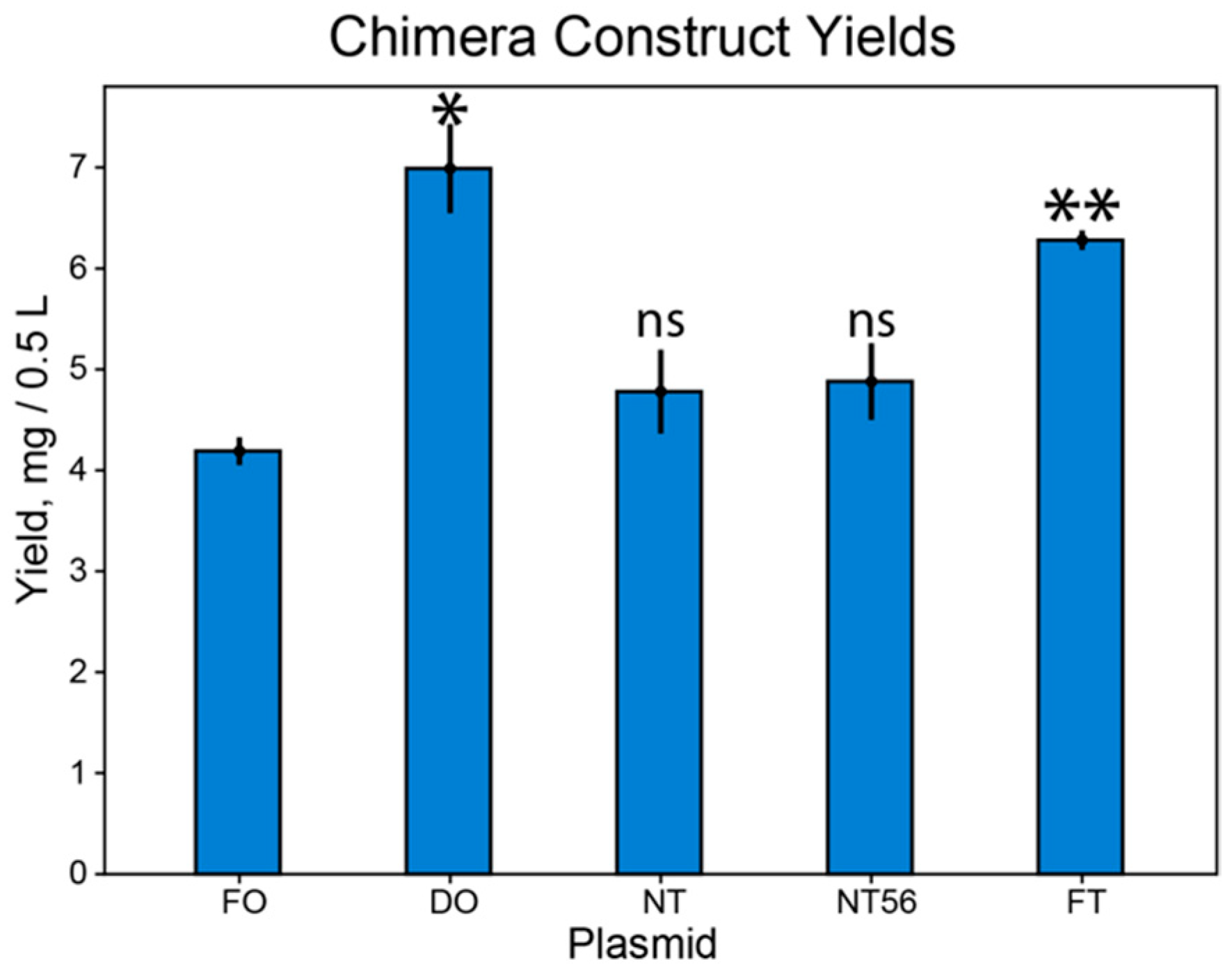
3.1. Codon Harmonization Increases Recombinant Protein Yield
3.2. Observed Yield Increase is Not Equal
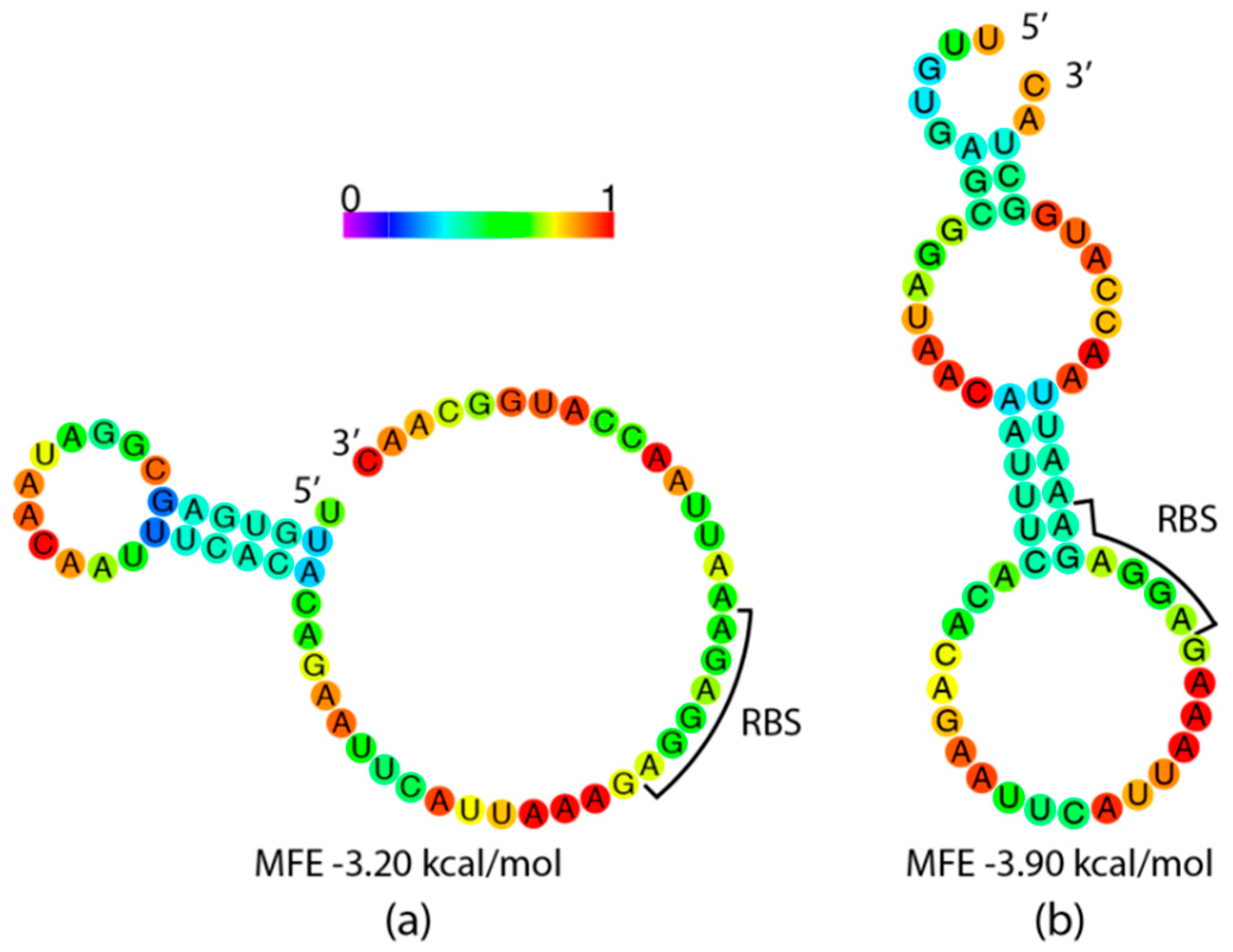
3.3. 5′ mRNA Secondary Structure as a Potential Contributing Factor in Observed Yield Inequality
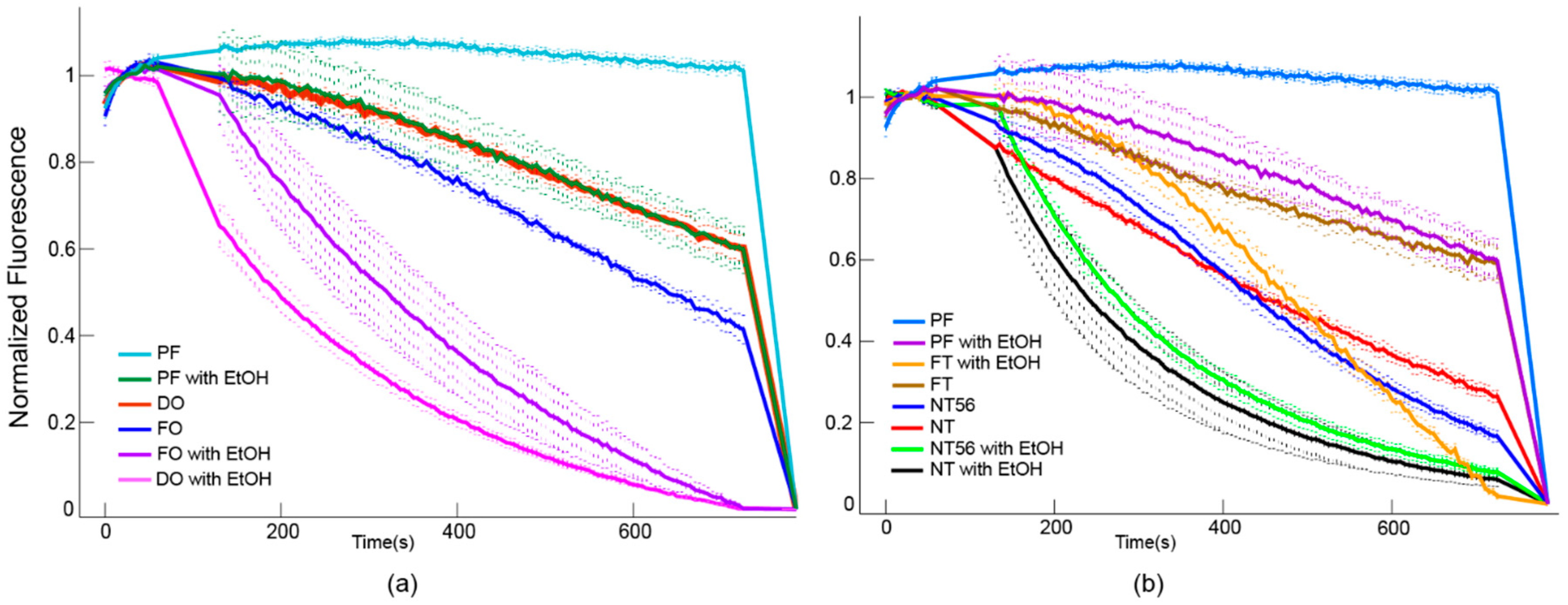
3.4. The Chimera is Biologically Active
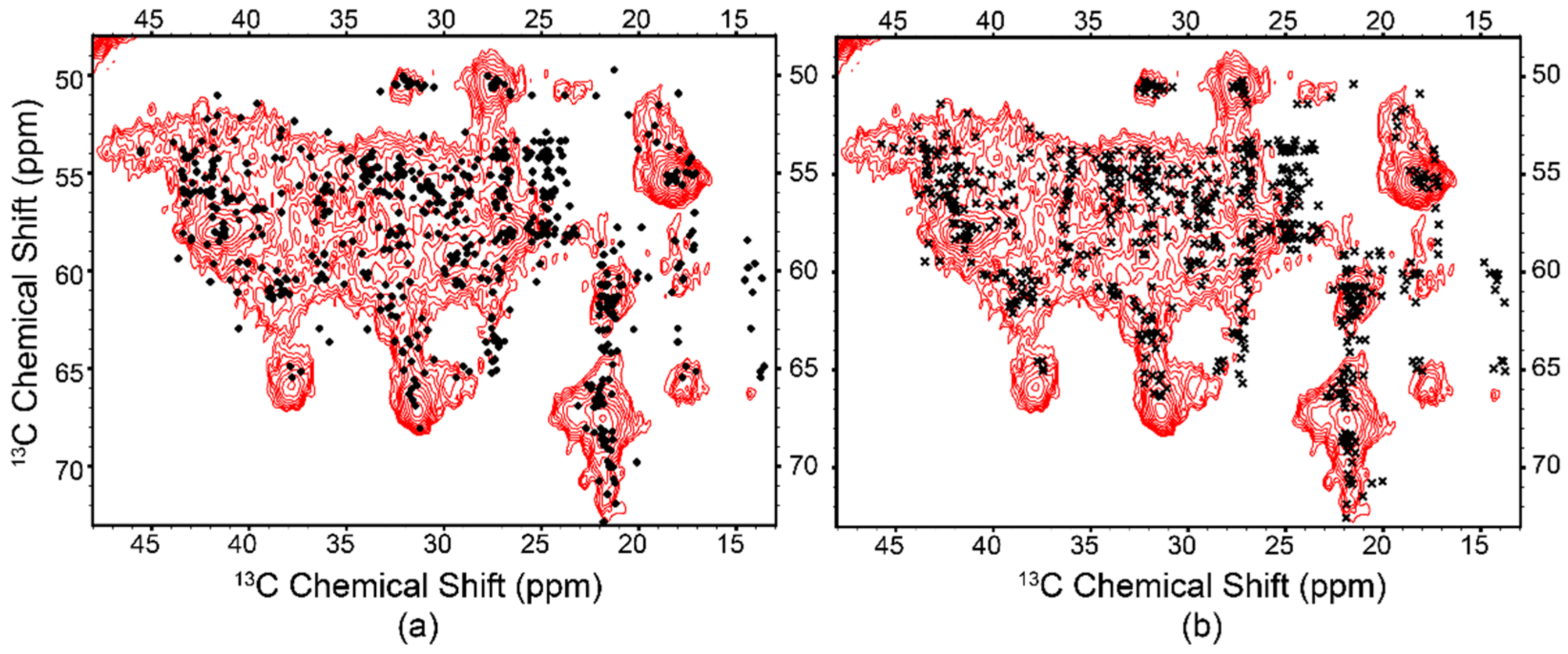
3.5. SSNMR Confirms Folded Protein in the Lipid Bilayer
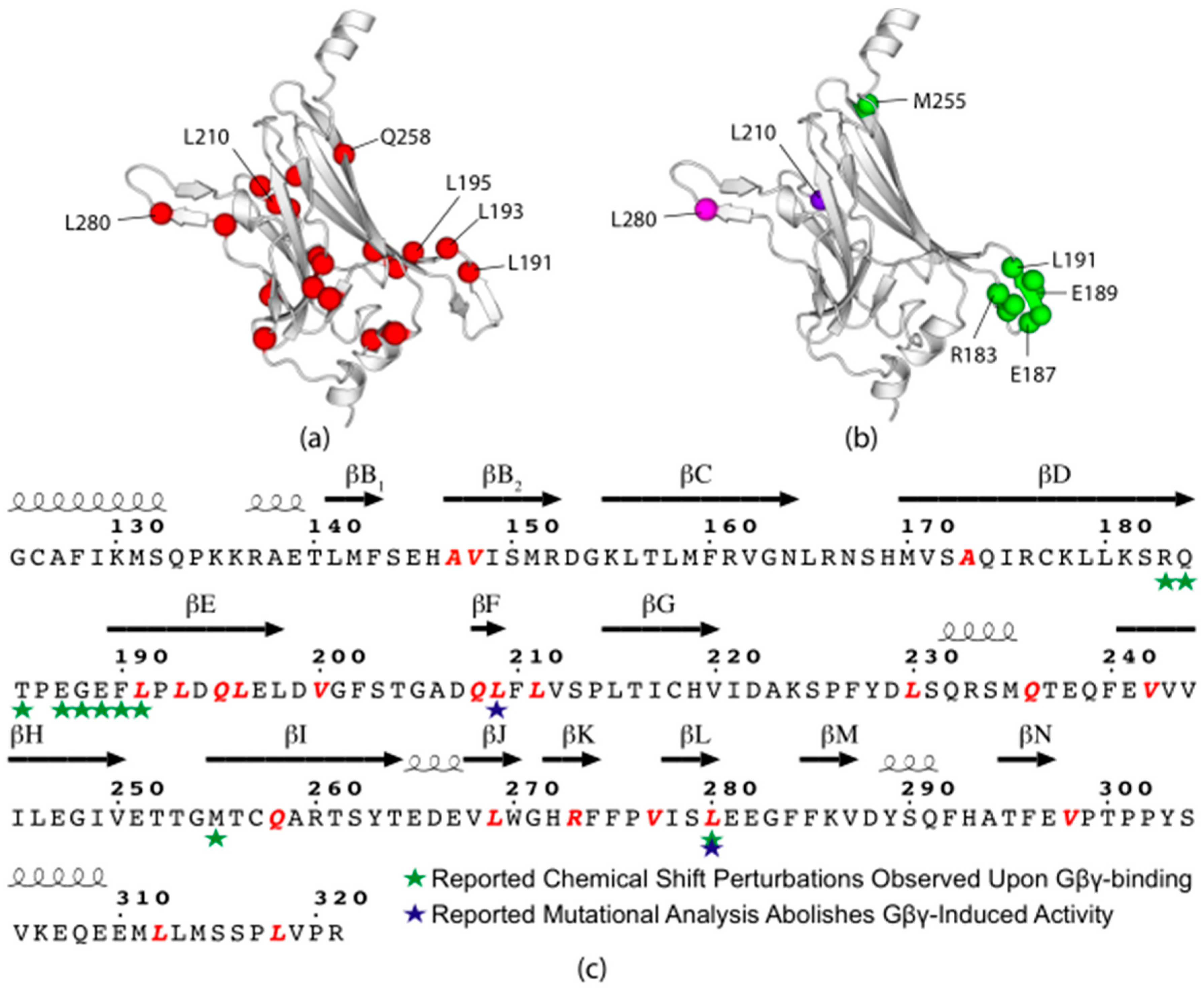
3.6. Rare Codons in the Eukaryotic Gating Domain
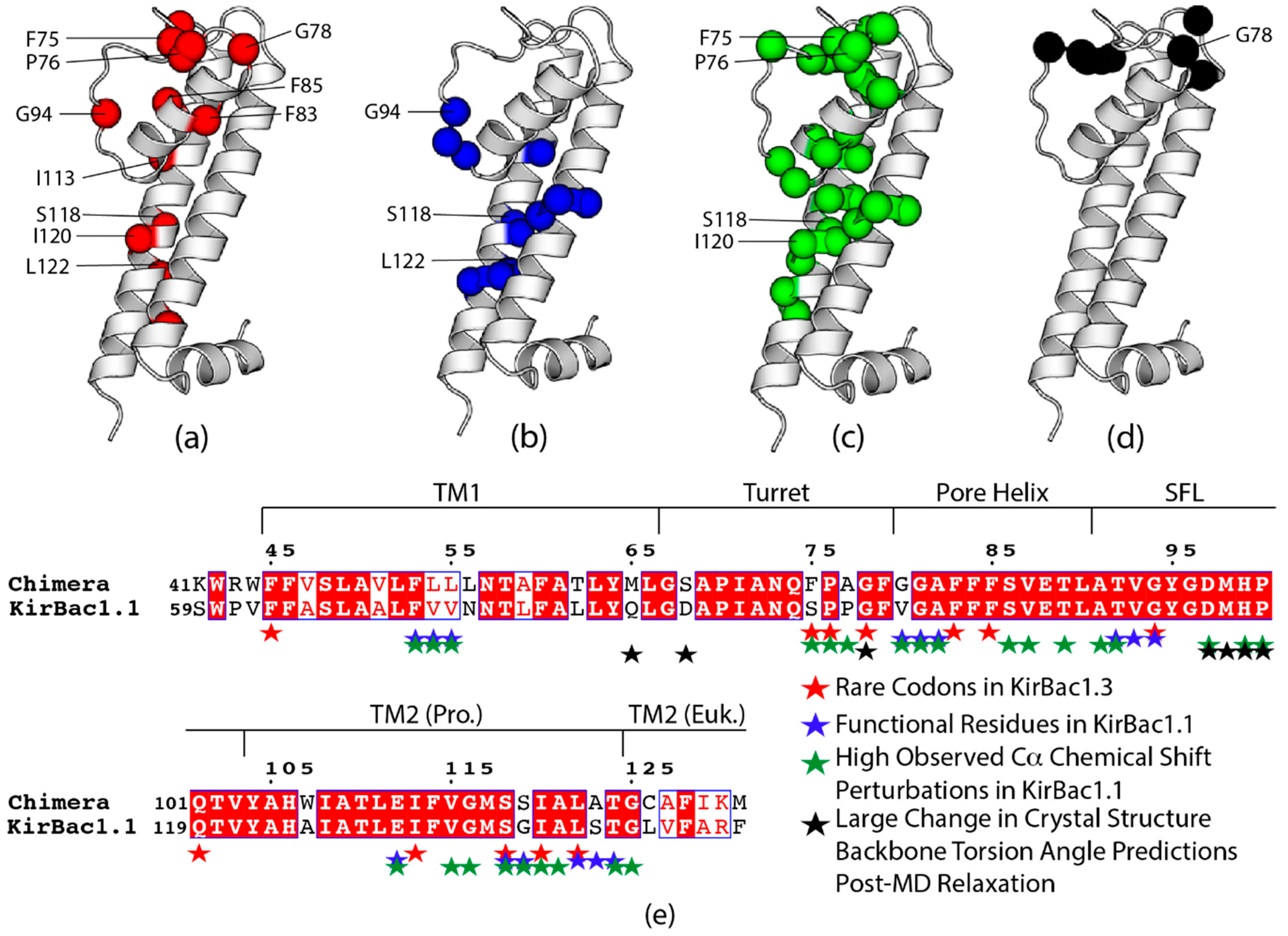
3.7. Rare Codons in the Prokaryotic Transmembrane Region
4. Discussion
4.1. Translation Initiation and Elongation as a Two-Step Model of Protein Synthesis
4.2. Other Variables as Co-Contributors to Yield Inequality
4.3. Structural Significance of Rare Codons
4.4. Considerations in Recombinant Gene Design
4.5. Codon Harmonization as an Easy-to-Implement Technique with Applications in Structural Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Liu, C.; Fan, Y.; Munro, R.A.; Ladizhansky, V.; Brown, L.S.; Wang, S. Sparse (13)C labelling for solid-state NMR studies of P. pastoris expressed eukaryotic seven-transmembrane proteins. J. Biomol. NMR 2016, 65, 7–13. [Google Scholar] [CrossRef]
- Fan, Y.; Emami, S.; Munro, R.; Ladizhansky, V.; Brown, L.S. Isotope labeling of eukaryotic membrane proteins in yeast for solid-state NMR. Methods Enzymol. 2015, 565, 193–212. [Google Scholar] [CrossRef]
- Ali, R.; Clark, L.D.; Zahm, J.A.; Lemoff, A.; Ramesh, K.; Rosenbaum, D.M.; Rosen, M.K. Improved strategy for isoleucine (1)H/(13)C methyl labeling in Pichia pastoris. J. Biomol. NMR 2019, 73, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.; Dikiy, I.; Rosenbaum, D.M.; Gardner, K.H. On the use of Pichia pastoris for isotopic labeling of human GPCRs for NMR studies. J. Biomol. NMR 2018, 71, 203–211. [Google Scholar] [CrossRef]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.W.; Oh, E.; Weissman, J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 2012, 484, 538–541. [Google Scholar] [CrossRef]
- Elf, J.; Ehrenberg, M. What makes ribosome-mediated transcriptional attenuation sensitive to amino acid limitation? PLoS Comput. Biol. 2005, 1, e2. [Google Scholar] [CrossRef] [Green Version]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, J.R.; Stansfield, I. Halting a cellular production line: Responses to ribosomal pausing during translation. Biol. Cell 2007, 99, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninio, J. Ribosomal kinetics and accuracy: Sequence engineering to the rescue. J. Mol. Biol. 2012, 422, 325–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, P.S.; Siller, E.; Anderson, J.F.; Barral, J.M. Silent Substitutions Predictably Alter Translation Elongation Rates and Protein Folding Efficiencies. J. Mol. Biol. 2012, 422, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siller, E.; DeZwaan, D.C.; Anderson, J.F.; Freeman, B.C.; Barral, J.M. Slowing bacterial translation speed enhances eukaryotic protein folding efficiency. J. Mol. Biol. 2010, 396, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Buhr, F.; Jha, S.; Thommen, M.; Mittelstaet, J.; Kutz, F.; Schwalbe, H.; Rodnina, M.V.; Komar, A.A. Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol. Cell 2016, 61, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quax, T.E.; Claassens, N.J.; Soll, D.; van der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.; Wright, G.; Emrich, S.; Clark, P.L. %MinMax: A versatile tool for calculating and comparing synonymous codon usage and its impact on protein folding. Protein Sci. 2018, 27, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angov, E.; Hillier, C.J.; Kincaid, R.L.; Lyon, J.A. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE 2008, 3, e2189. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.; Minshull, J.; Govindarajan, S.; Ness, J.; Villalobos, A.; Welch, M. Engineering genes for predictable protein expression. Protein Expr. Purif. 2012, 83, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Chevance, F.F.; Le Guyon, S.; Hughes, K.T. The effects of codon context on in vivo translation speed. PLoS Genet. 2014, 10, e1004392. [Google Scholar] [CrossRef] [Green Version]
- Angov, E.; Legler, P.M.; Mease, R.M. Adjustment of codon usage frequencies by codon harmonization improves protein expression and folding. Methods Mol. Biol. 2011, 705, 1–13. [Google Scholar] [CrossRef]
- Mignon, C.; Mariano, N.; Stadthagen, G.; Lugari, A.; Lagoutte, P.; Donnat, S.; Chenavas, S.; Perot, C.; Sodoyer, R.; Werle, B. Codon harmonization - going beyond the speed limit for protein expression. FEBS Lett. 2018, 592, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Owen, G.R.; Achilonu, I.; Dirr, H.W. High yield purification of JNK1beta1 and activation by in vitro reconstitution of the MEKK1-->MKK4-->JNK MAPK phosphorylation cascade. Protein Expr. Purif. 2013, 87, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Rehbein, P.; Berz, J.; Kreisel, P.; Schwalbe, H. “CodonWizard”—An intuitive software tool with graphical user interface for customizable codon optimization in protein expression efforts. Protein Expr. Purif. 2019, 160, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. Embo J. 2007, 26, 4005–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whorton, M.R.; MacKinnon, R. Crystal Structure of the Mammalian GIRK2 K+ Channel and Gating Regulation by G Proteins, PIP2, and Sodium. Cell 2011, 147, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stansfeld, P.J.; Hopkinson, R.; Ashcroft, F.M.; Sansom, M.S. PIP(2)-binding site in Kir channels: Definition by multiscale biomolecular simulations. Biochemistry 2009, 48, 10926–10933. [Google Scholar] [CrossRef] [Green Version]
- Aryal, P.; Dvir, H.; Choe, S.; Slesinger, P.A. A discrete alcohol pocket involved in GIRK channel activation. Nat. Neurosci. 2009, 12, 988–995. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Hausherr, A.; Ferbitz, L.; Schodl, T.; Heitzer, M.; Hegemann, P. Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant. Mol. Biol. 2004, 55, 869–881. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Clarke, T.F., 4th; Clark, P.L. Rare codons cluster. PLoS ONE 2008, 3, e3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Borcik, C.G.; Versteeg, D.B.; Wylie, B.J. An Inward-Rectifier Potassium Channel Coordinates the Properties of Biologically Derived Membranes. Biophys J. 2019, 116, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Bhate, M.P.; Wylie, B.J.; Thompson, A.; Tian, L.; Nimigean, C.; McDermott, A.E. Preparation of uniformly isotope labeled KcsA for solid state NMR: Expression, purification, reconstitution into liposomes and functional assay. Protein Expr. Purif. 2013, 91, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Gor’kov, P.L.; Chekmenev, E.Y.; Li, C.G.; Cotten, M.; Buffy, J.J.; Traaseth, N.J.; Veglia, G.; Brey, W.W. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J. Magn. Reson. 2007, 185, 77–93. [Google Scholar] [CrossRef]
- McNeill, S.A.; Gor’kov, P.L.; Shetty, K.; Brey, W.W.; Long, J.R. A low-E magic angle spinning probe for biological solid state NMR at 750 MHz. J. Magn. Reson. 2009, 197, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Takegoshi, K.; Nakamura, S.; Terao, T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001, 344, 631–637. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef]
- Orekhov, V.Y.; Ibraghimov, I.; Billeter, M. Optimizing resolution in multidimensional NMR by three-way decomposition. J. Biomol. NMR 2003, 27, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Luan, T.; Jaravine, V.; Yee, A.; Arrowsmith, C.H.; Orekhov, V.Y. Optimization of resolution and sensitivity of 4D NOESY using multi-dimensional decomposition. J. Biomol. NMR 2005, 33, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jaravine, V.; Ibraghimov, I.; Orekhov, V.Y. Removal of a time barrier for high-resolution multidimensional NMR spectroscopy. Nat. Methods 2006, 3, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Liu, Y.; Ginzinger, S.W.; Wishart, D.S. SHIFTX2: Significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Gradmann, S.; Ader, C.; Heinrich, I.; Nand, D.; Dittmann, M.; Cukkemane, A.; van Dijk, M.; Bonvin, A.M.J.J.; Engelhard, M.; Baldus, M. Rapid prediction of multi-dimensional NMR data sets. J. Biomol. NMR 2012, 54, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Borujeni, A.E.; Cetnar, D.; Farasat, I.; Smith, A.; Lundgren, N.; Salis, H.M. Precise quantification of translation inhibition by mRNA structures that overlap with the ribosomal footprint in N-terminal coding sequences. Nucleic Acids Res. 2017, 45, 5437–5448. [Google Scholar] [CrossRef]
- Su, Z.W.; Brown, E.C.; Wang, W.W.; MacKinnon, R. Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc. Natl. Acad. Sci. USA 2016, 113, 5748–5753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Hubalewska, M.; Ignatova, Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 2009, 16, 274–280. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Logothetis, D.E.; Cui, M. The molecular mechanism by which PIP(2) opens the intracellular G-loop gate of a Kir3.1 channel. Biophys. J. 2012, 102, 2049–2059. [Google Scholar] [CrossRef] [Green Version]
- Whorton, M.R.; MacKinnon, R. X-ray structure of the mammalian GIRK2-beta gamma G-protein complex. Nature 2013, 498, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Toyama, Y.; Kano, H.; Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Structural basis for the ethanol action on G-protein-activated inwardly rectifying potassium channel 1 revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2018, 115, 3858–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokogawa, M.; Osawa, M.; Takeuchi, K.; Mase, Y.; Shimada, I. NMR analyses of the Gbetagamma binding and conformational rearrangements of the cytoplasmic pore of G protein-activated inwardly rectifying potassium channel 1 (GIRK1). J. Biol. Chem. 2011, 286, 2215–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Zhang, H.L.; Mirshahi, T.; Logothetis, D.E. Identification of a potassium channel site that interacts with G protein beta gamma subunits to mediate agonist-induced signaling. J. Biol. Chem. 1999, 274, 12517–12524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Yan, X.; Zhang, H.; Mirshahi, T.; Jin, T.; Huang, A.; Logothetis, D.E. Identification of critical residues controlling G protein-gated inwardly rectifying K(+) channel activity through interactions with the beta gamma subunits of G proteins. J. Biol. Chem. 2002, 277, 6088–6096. [Google Scholar] [CrossRef] [Green Version]
- Amani, R.; Borcik, C.G.; Khan, N.H.; Versteeg, D.B.; Yekefallah, M.; Do, H.Q.; Coats, H.R.; Wylie, B.J. Conformational changes upon gating of KirBac1.1 into an open-activated state revealed by solid-state NMR and functional assays. Proc. Natl. Acad. Sci. USA 2020, 117, 2938–2947. [Google Scholar] [CrossRef] [Green Version]
- Bentele, K.; Saffert, P.; Rauscher, R.; Ignatova, Z.; Bluthgen, N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013, 9, 675. [Google Scholar] [CrossRef]
- Tuller, T.; Waldman, Y.Y.; Kupiec, M.; Ruppin, E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA 2010, 107, 3645–3650. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Kumar, A.; Kaur, J. Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. Int. J. Biol Macromol. 2018, 106, 803–822. [Google Scholar] [CrossRef]
- Boel, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.H.; Su, M.; Luff, J.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Carrio, M.M.; Cubarsi, R.; Villaverde, A. Fine architecture of bacterial inclusion bodies. FEBS Lett. 2000, 471, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Rosano, G.L.; Ceccarelli, E.A. Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb. Cell Fact. 2009, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, G.N.; Clark, P.L. Quality over quantity: Optimizing co-translational protein folding with non-’optimal’ synonymous codons. Curr. Opin. Struct. Biol. 2016, 38, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortazzo, P.; Cervenansky, C.; Marin, M.; Reiss, C.; Ehrlich, R.; Deana, A. Silent mutations affect in vivo protein folding in Escherichia coli. Biochem. Bioph. Res. Commun. 2002, 293, 537–541. [Google Scholar] [CrossRef]
- Makrides, S.C. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 1996, 60, 512–538. [Google Scholar] [CrossRef] [Green Version]
- Pechmann, S.; Frydman, J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 2013, 20, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Aalst, E.; Yekefallah, M.; Mehta, A.K.; Eason, I.; Wylie, B. Codon Harmonization of a Kir3.1-KirBac1.3 Chimera for Structural Study Optimization. Biomolecules 2020, 10, 430. https://doi.org/10.3390/biom10030430
Van Aalst E, Yekefallah M, Mehta AK, Eason I, Wylie B. Codon Harmonization of a Kir3.1-KirBac1.3 Chimera for Structural Study Optimization. Biomolecules. 2020; 10(3):430. https://doi.org/10.3390/biom10030430
Chicago/Turabian StyleVan Aalst, Evan, Maryam Yekefallah, Anil K. Mehta, Isaac Eason, and Benjamin Wylie. 2020. "Codon Harmonization of a Kir3.1-KirBac1.3 Chimera for Structural Study Optimization" Biomolecules 10, no. 3: 430. https://doi.org/10.3390/biom10030430
APA StyleVan Aalst, E., Yekefallah, M., Mehta, A. K., Eason, I., & Wylie, B. (2020). Codon Harmonization of a Kir3.1-KirBac1.3 Chimera for Structural Study Optimization. Biomolecules, 10(3), 430. https://doi.org/10.3390/biom10030430