Fluorogenic and Bioorthogonal Modification of RNA Using Photoclick Chemistry


Abstract
:1. Introduction
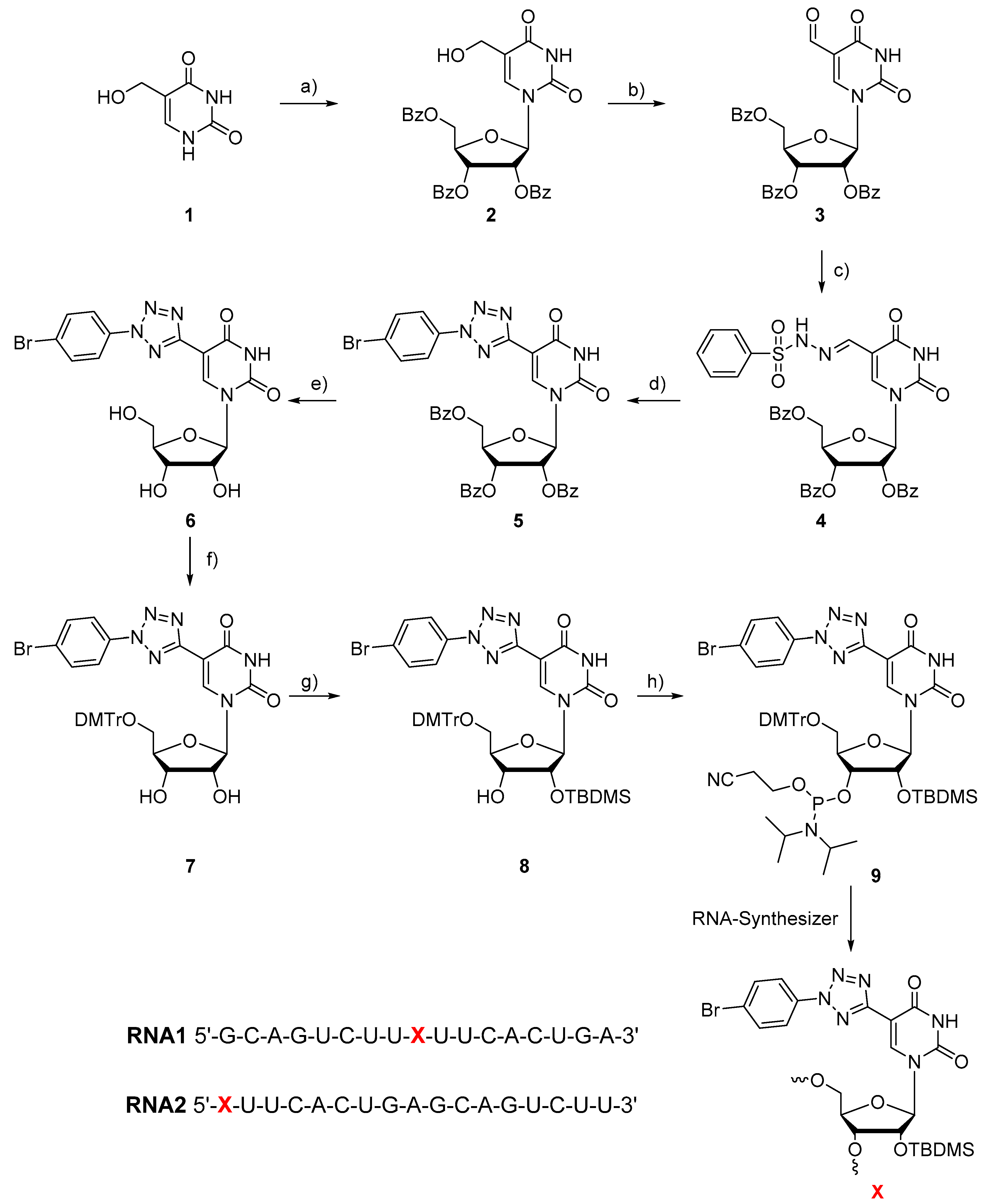
2. Materials and Methods
General
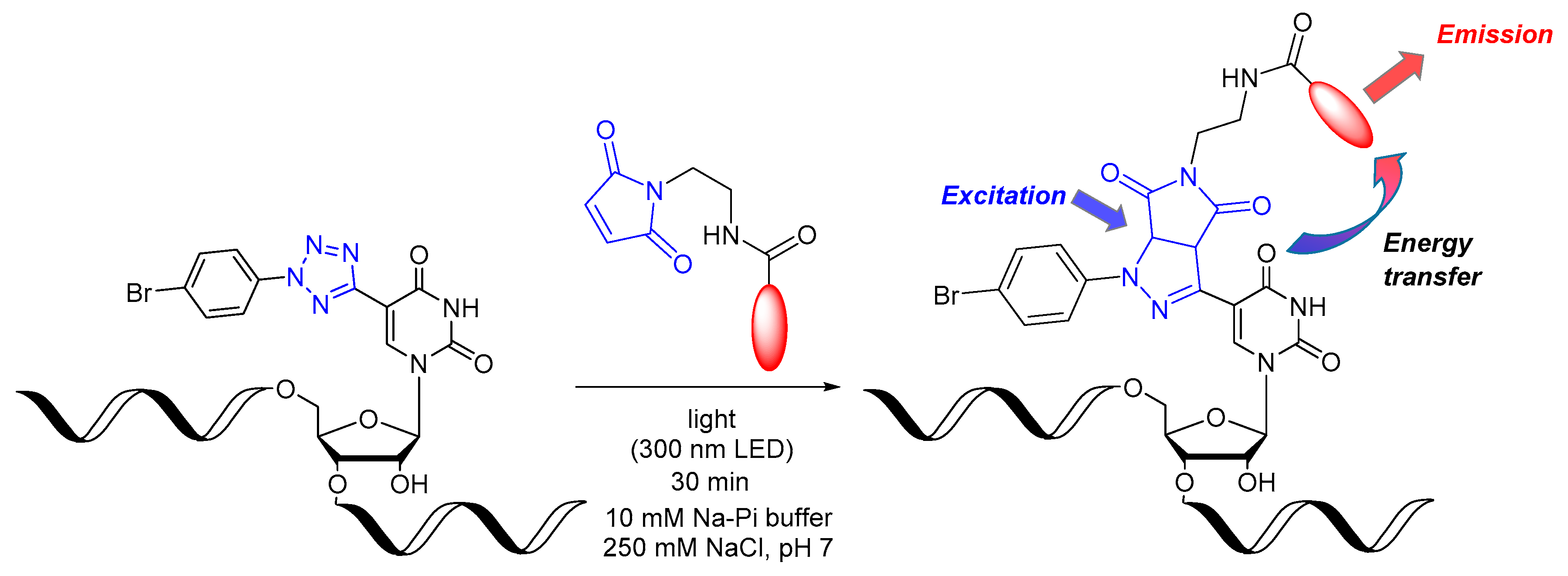
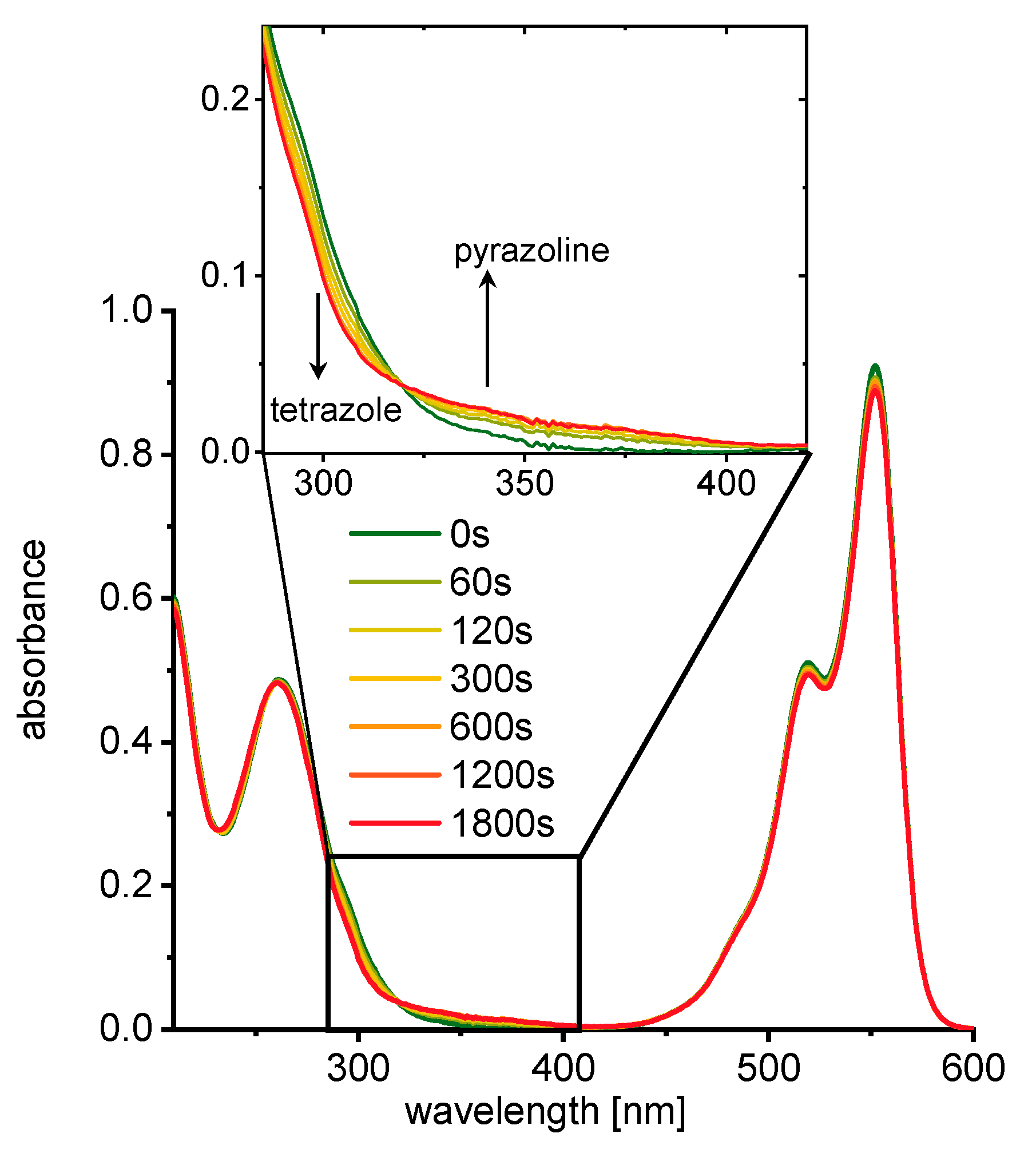
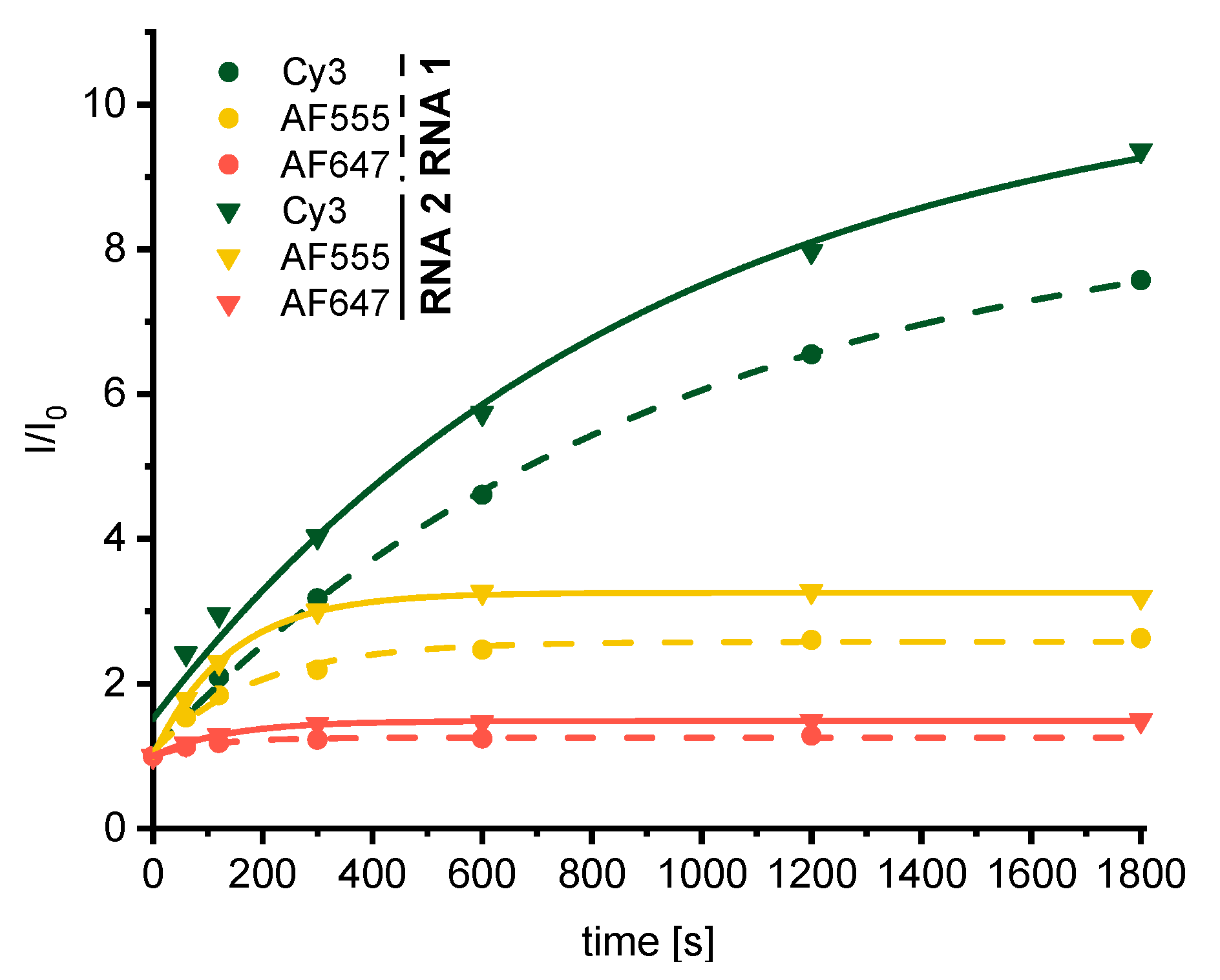
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Row, R.D.; Prescher, J.A. Constructing New Bioorthogonal Reagents and Reactions. Accounts Chem. Res. 2018, 51, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Shieh, P.; Bertozzi, C.R. Design strategies for bioorthogonal smart probes. Org. Biomol. Chem. 2014, 12, 9307–9320. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, N.K. The Future of Bioorthogonal Chemistry. ACS Central Sci. 2018, 4, 952–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Hong, V.; Steinmetz, N.F.; Manchester, M.; Finn, M.G. Labeling Live Cells by Copper-Catalyzed Alkyne−Azide Click Chemistry. Bioconjugate Chem. 2010, 21, 1912–1916. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, J.; Chen, P.R. Transition metal-mediated bioorthogonal protein chemistry in living cells. Chem. Soc. Rev. 2014, 43, 6511–6526. [Google Scholar] [CrossRef]
- Kennedy, D.; McKay, C.S.; Legault, M.C.B.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular Consequences of Copper Complexes Used To Catalyze Bioorthogonal Click Reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef]
- Devaraj, N.K.; Weissleder, R.; Hilderbrand, S.A. Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chem. 2008, 19, 2297–2299. [Google Scholar] [CrossRef] [Green Version]
- Lang, K.; Mayer, S. Tetrazines in Inverse-Electron-Demand Diels–Alder Cycloadditions and Their Use in Biology. Synthesis 2016, 49, 830–848. [Google Scholar] [CrossRef]
- Kamber, D.N.; Liang, Y.; Blizzard, R.J.; Liu, F.; Mehl, R.A.; Houk, K.N.; Prescher, J.A. 1,2,4-Triazines Are Versatile Bioorthogonal Reagents. J. Am. Chem. Soc. 2015, 137, 8388–8391. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Chin, J.W. Bioorthogonal Reactions for Labeling Proteins. ACS Chem. Biol. 2014, 9, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.K.V.; Lin, Q. Photoinducible Bioorthogonal Chemistry: A Spatiotemporally Controllable Tool to Visualize and Perturb Proteins in Live Cells. Accounts Chem. Res. 2011, 44, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Nadler, A.; Schultz, C. The Power of Fluorogenic Probes. Angew. Chem. Int. Ed. 2013, 52, 2408–2410. [Google Scholar] [CrossRef]
- Wu, H.; Devaraj, N.K. Advances in Tetrazine Bioorthogonal Chemistry Driven by the Synthesis of Novel Tetrazines and Dienophiles. Accounts Chem. Res. 2018, 51, 1249–1259. [Google Scholar] [CrossRef]
- Wieczorek, A.; Werther, P.; Euchner, J.; Wombacher, R. Green- to far-red-emitting fluorogenic tetrazine probes - synthetic access and no-wash protein imaging inside living cells. Chem. Sci. 2017, 8, 1506–1510. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.; Wagenknecht, H.-A. Fluorogenic “photoclick”-type labelling of DNA using a Cy3 dye. Org. Biomol. Chem. 2018, 16, 7579–7582. [Google Scholar] [CrossRef]
- Hudson, R.H.; Wojciechowski, F. The detrimental effect of orotic acid substitution in the peptide nucleic acid strand on the stability of PNA2:NA triple helices. Can. J. Chem. 2005, 83, 1731–1740. [Google Scholar] [CrossRef]
- Clovis, J.S.; Eckell, A.; Huisgen, R.; Sustmann, R. 1.3-Dipolare Cycloadditionen, XXV. Der Nachweis des freien Diphenylnitrilimins als Zwischenstufe bei Cycloadditionen. Eur. J. Inorg. Chem. 1967, 100, 60–70. [Google Scholar] [CrossRef]
- Blasco, E.; Sugawara, Y.; Lederhose, P.; Blinco, J.P.; Kelterer, A.-M.; Barner-Kowollik, C. Understanding Reactivity Patterns in Light-Induced Nitrile Imine Mediated Tetrazole-Ene Cycloadditions. ChemPhotoChem 2017, 1, 159–163. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Krolikiewicz, K. Neue Katalysatoren für die Nucleosidsynthese. Angew. Chem. 1975, 87, 417. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Kakehi, A.; Tanaka, Y.; Ito, S.; Kondo, K. A Facile Synthesis of 2,5-Disubstituted Tetrazoles by the Reaction of Phenylsulfonylhydrazones with Arenediazonium Salts. Bull. Chem. Soc. Jpn. 1976, 49, 1920–1923. [Google Scholar]
- Hakimelahi, G.H.; Proba, Z.A.; Ogilvie, K.K. New catalysts and procedures for the dimethoxytritylation and selective silylation of ribonucleosides. Can. J. Chem. 1982, 60, 1106–1113. [Google Scholar] [CrossRef]
- Tridgett, M.; Moore-Kelly, C.; Duprey, J.-L.H.A.; Iturbe, L.O.; Tsang, C.W.; Little, H.A.; Sandhu, S.K.; Hicks, M.R.; Dafforn, T.R.; Rodger, A. Linear dichroism of visible-region chromophores using M13 bacteriophage as an alignment scaffold. RSC Adv. 2018, 8, 29535–29543. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye Adduct of | RNA1 | RNA2 |
|---|---|---|
| Cy3 | 27% | 31% (70%) a |
| AF555 | 78% | 84% |
| AF647 | 48% | 48% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krell, K.; Wagenknecht, H.-A. Fluorogenic and Bioorthogonal Modification of RNA Using Photoclick Chemistry. Biomolecules 2020, 10, 480. https://doi.org/10.3390/biom10030480
Krell K, Wagenknecht H-A. Fluorogenic and Bioorthogonal Modification of RNA Using Photoclick Chemistry. Biomolecules. 2020; 10(3):480. https://doi.org/10.3390/biom10030480
Chicago/Turabian StyleKrell, Katja, and Hans-Achim Wagenknecht. 2020. "Fluorogenic and Bioorthogonal Modification of RNA Using Photoclick Chemistry" Biomolecules 10, no. 3: 480. https://doi.org/10.3390/biom10030480
APA StyleKrell, K., & Wagenknecht, H. -A. (2020). Fluorogenic and Bioorthogonal Modification of RNA Using Photoclick Chemistry. Biomolecules, 10(3), 480. https://doi.org/10.3390/biom10030480