1H-Imidazole-2,5-Dicarboxamides as NS4A Peptidomimetics: Identification of a New Approach to Inhibit HCV-NS3 Protease

 ,
,  ,
, 
 , ,
, ,
Abstract
:
1. Introduction
2. Results
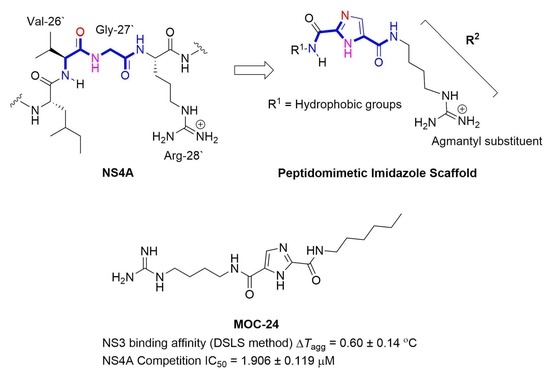
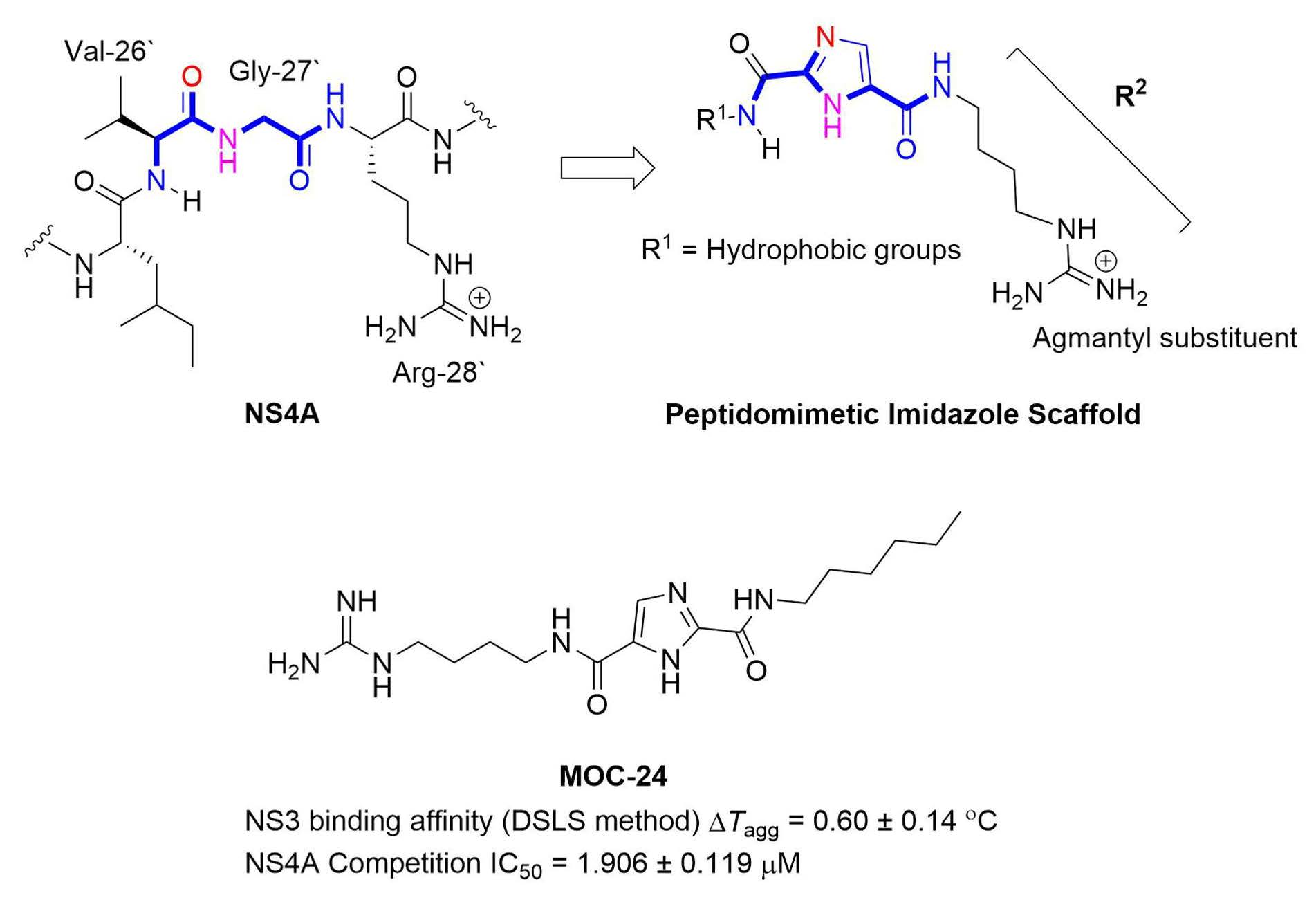
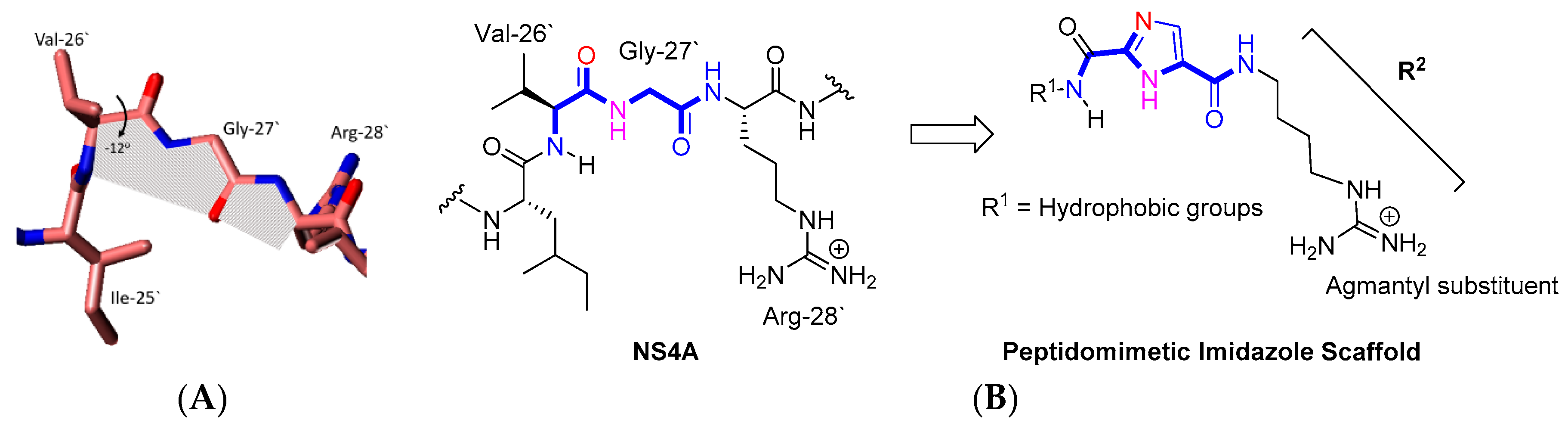
2.1. Rationale and Design
2.2. Synthesis of 1H-Imidazole-2,5-Dicarboxamide Derivatives
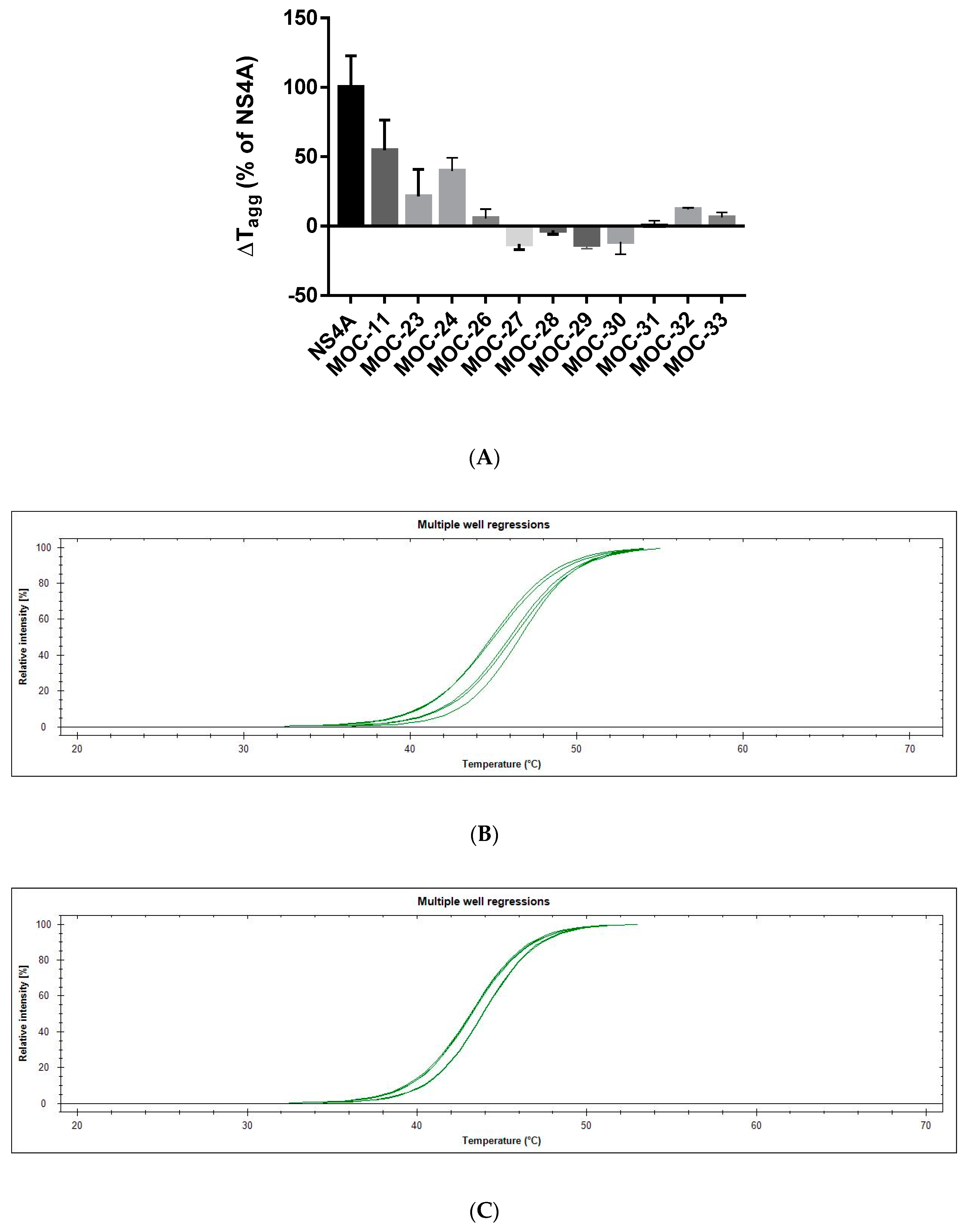
2.3. Comparative Binding Evaluation of MOC Compounds Using Differential Static Light Scattering (DSLS)
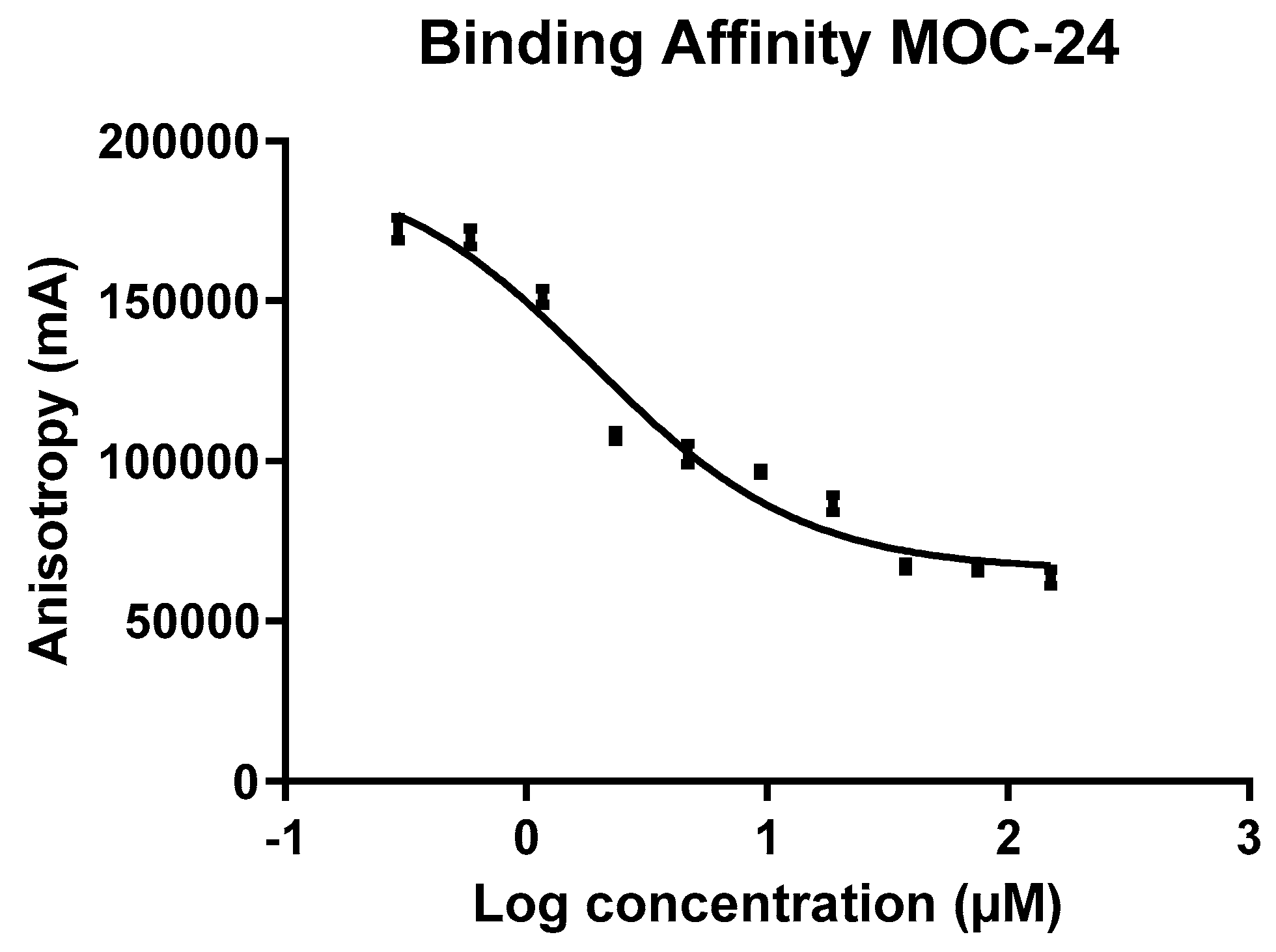
2.4. Fluorescence Anisotropy (FA) Competition Assay of MOC Compounds with NS4A
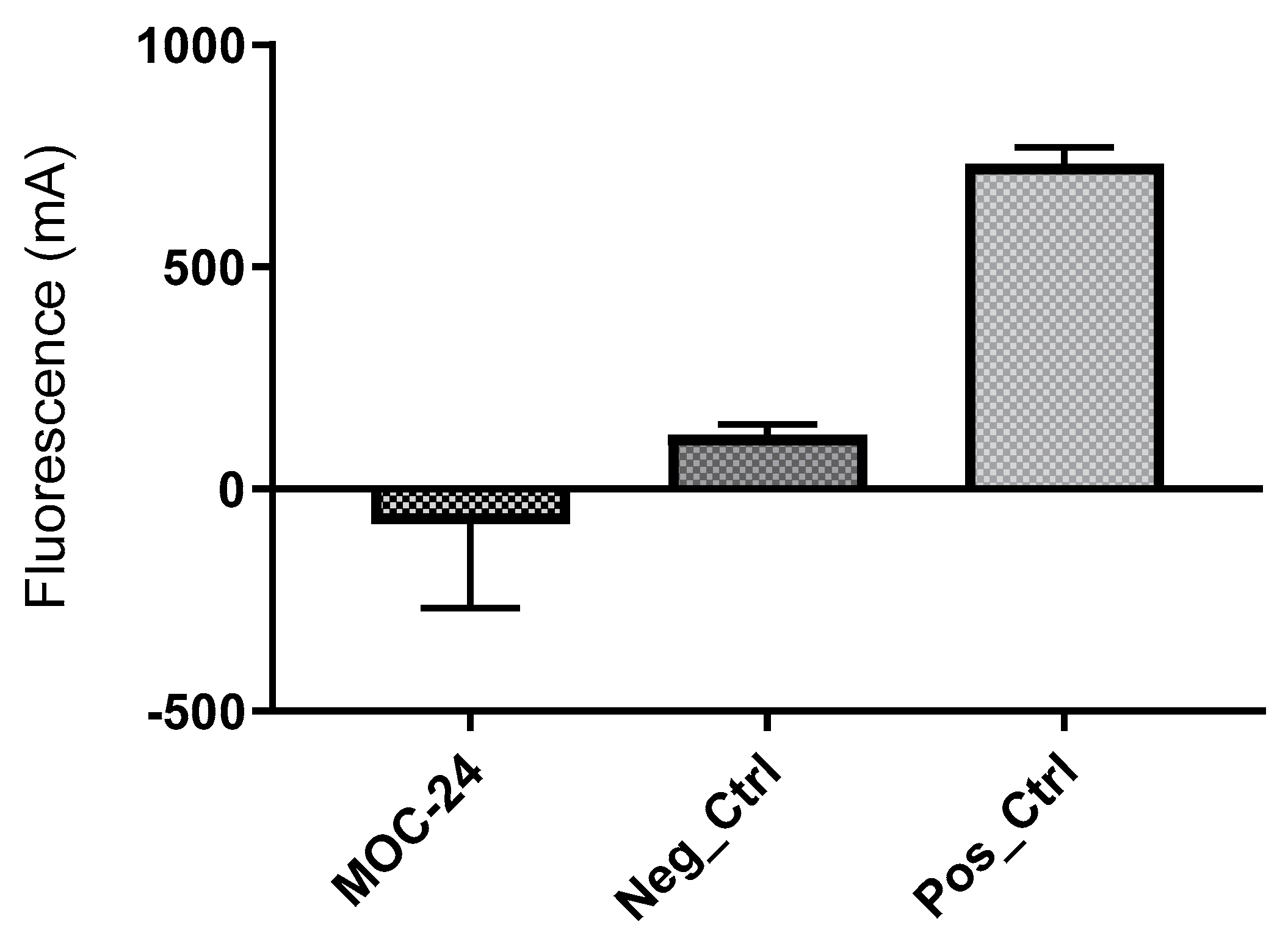
2.5. Enzyme Inhibition Assay
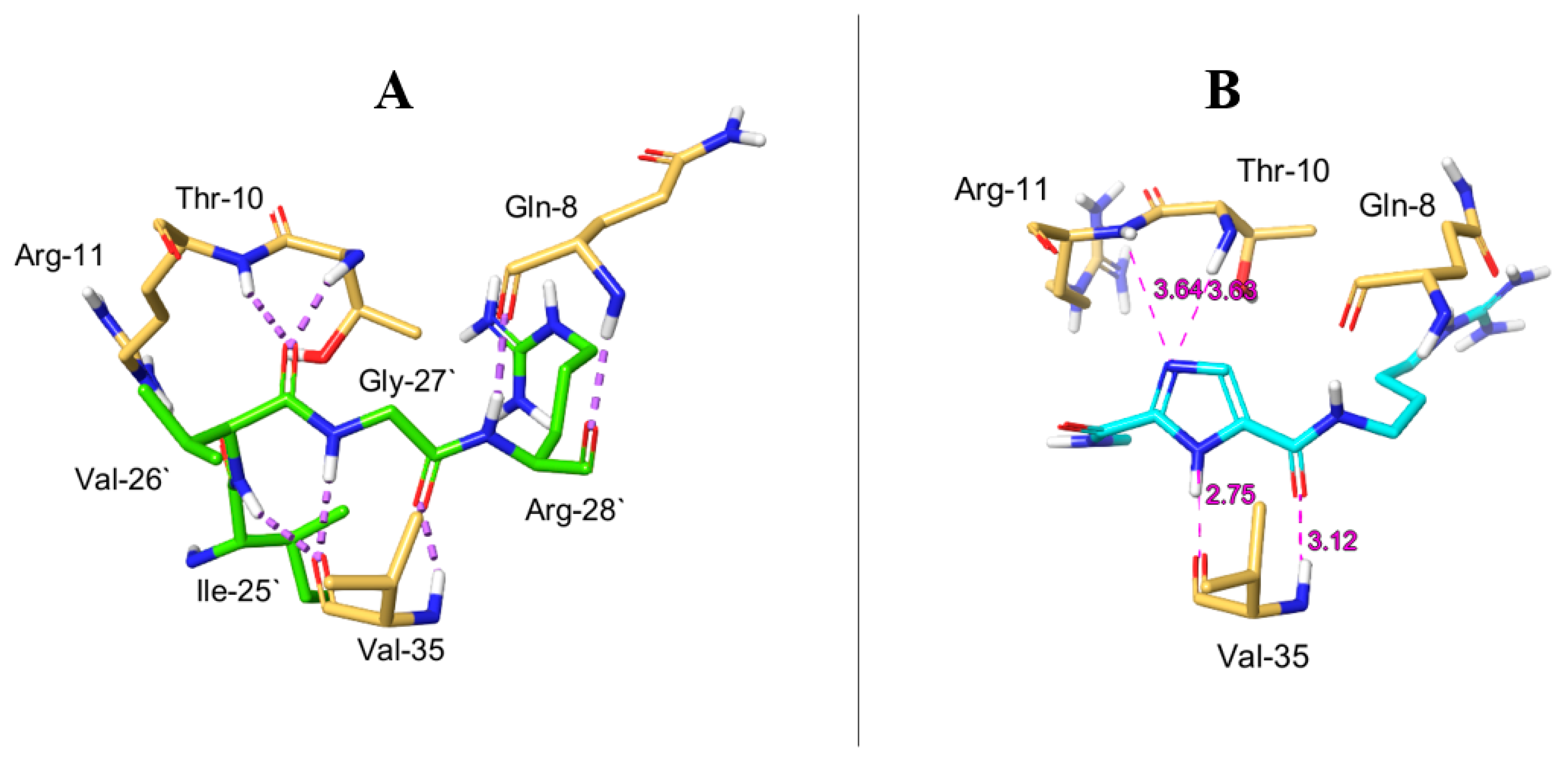
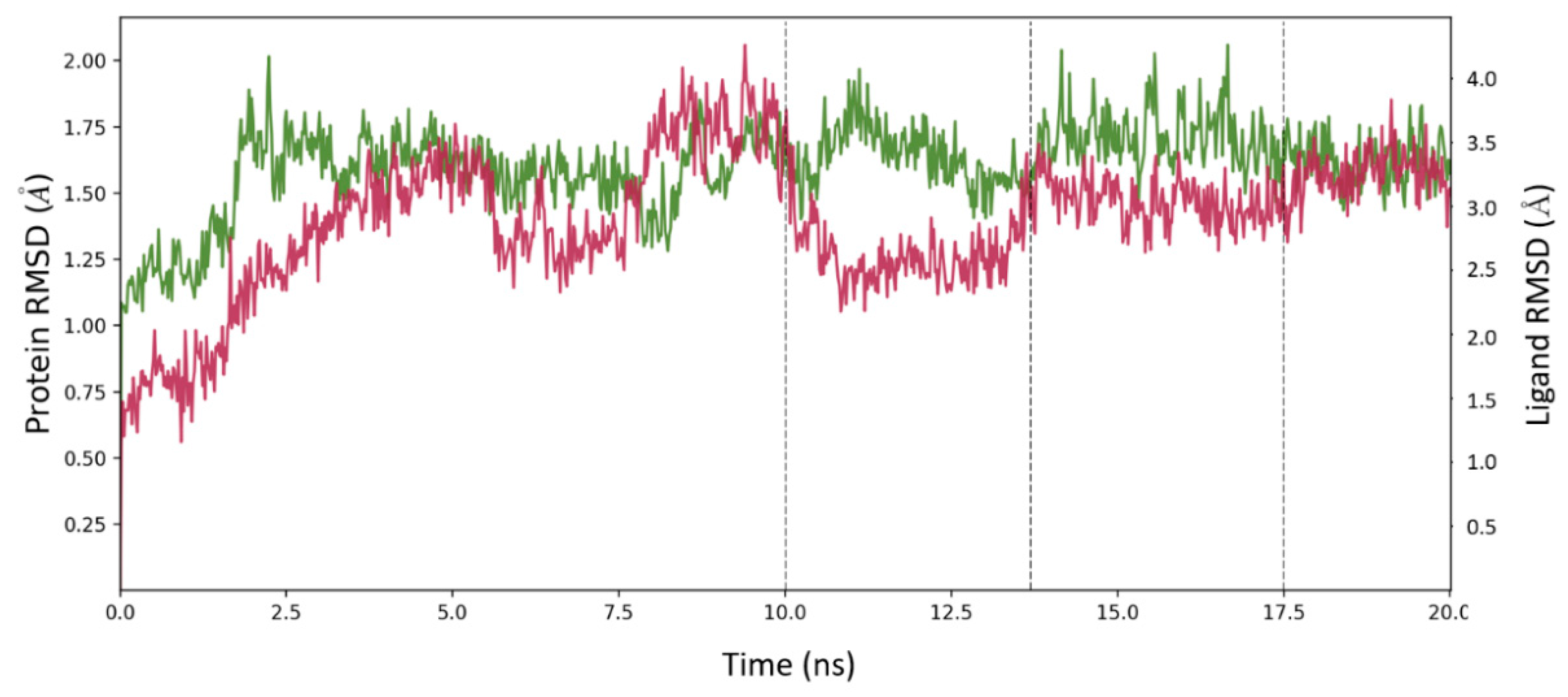
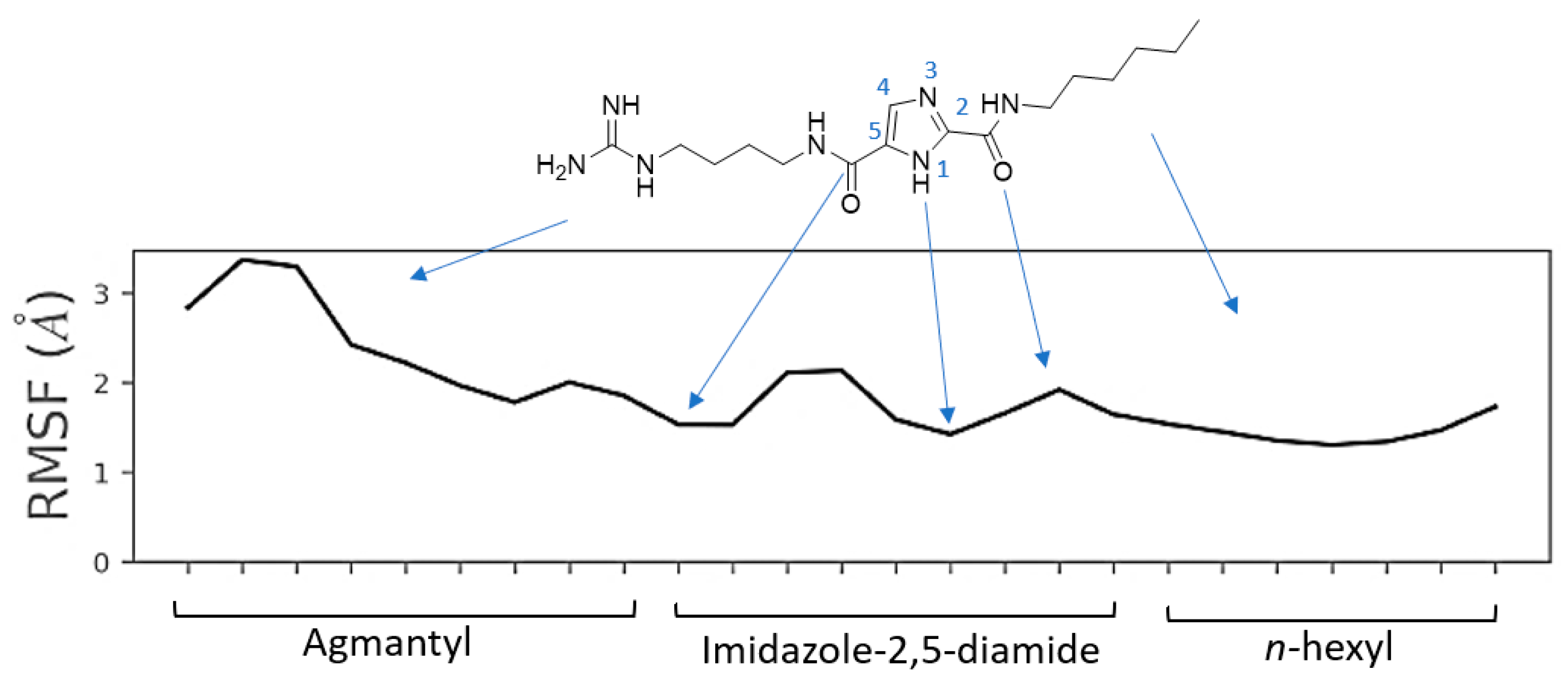
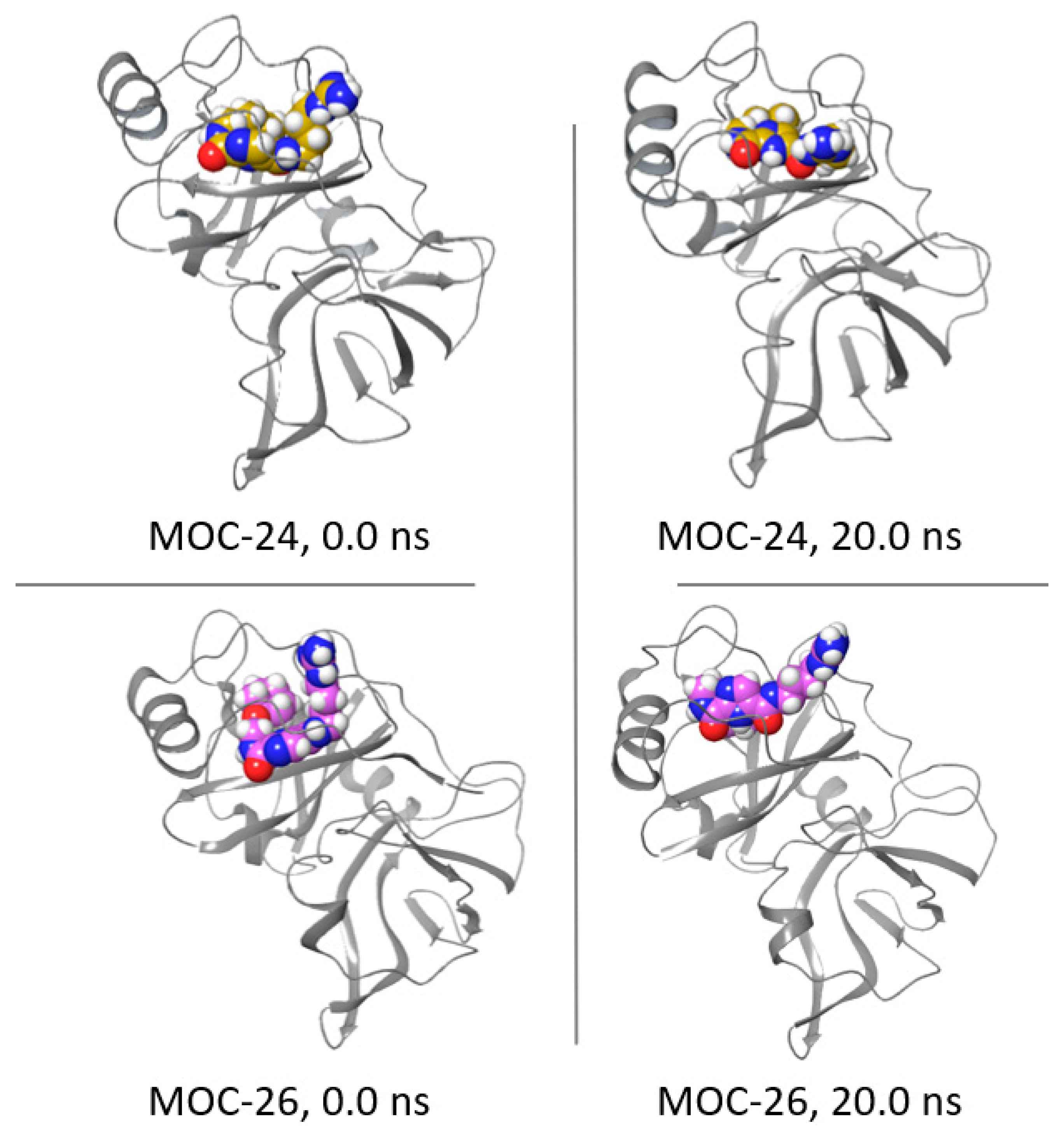
2.6. Molecular Dynamics Simulation for MOC-24 and MOC-26
3. Discussion and Conclusions
4. Experimental
4.1. Chemical Synthesis
4.1.1. Synthesis of 5-(Tert-Butyl) 2-Ethyl 1H-Imidazole-2,5-Dicarboxylate (3)
4.1.2. Synthesis of 2-(Ethoxycarbonyl)-1H-Imidazole-4-Carboxylic Acid (4)
4.1.3. General Procedure for the Synthesis of Ethyl 4-(N-Substituted Carbamoyl)-1H-Imidazole-2-Carboxylate (5a‒5c)
4.1.4. General Procedure for Synthesis of 6a–6c
N5-(2-(Methylamino)-2-oxoethyl)-N2-(n-pentyl)-1H-imidazole-2,5-dicarboxamide (MOC-11)
N2-(n-Hexyl)-N5-(pyridin-2-ylmethyl)-1H-imidazole-2,5-dicarboxamide (MOC-33)
N5-(4-Guanidinobutyl)-N2-(n-pentyl)-1H-imidazole-2,5-dicarboxamide (MOC-23)
N5-(4-Guanidinobutyl)-N2-(n-hexyl)-1H-imidazole-2,5-dicarboxamide (MOC-24)
N2-(2-(Cyclohexyloxy)ethyl)-N5-(4-guanidinobutyl)-1H-imidazole-2,5-dicarboxamide (MOC-26)
N2-(3-(Cyclohexyloxy)propyl)-N5-(4-guanidinobutyl)-1H-imidazole-2,5-dicarboxamide (MOC-27)
N5-(4-Guanidinobutyl)-N2-(2-phenoxyethyl)-1H-imidazole-2,5-dicarboxamide (MOC-28)
N5-(4-Guanidinobutyl)-N2-(3-phenoxypropyl)-1H-imidazole-2,5-dicarboxamide (MOC-29)
N5-(4-Guanidinobutyl)-N2-(4-isopropoxybutyl)-1H-imidazole-2,5-dicarboxamide (MOC-30)
N5-(4-Guanidinobutyl)-N2-(6-methylheptyl)-1H-imidazole-2,5-dicarboxamide (MOC-31)
N5-(4-guanidinobutyl)-N2-(3-methylpentyl)-1H-imidazole-2,5-dicarboxamide (MOC-32)
4.2. Biological Screening
4.2.1. NS3 Protein
NS3 Constructs
NS3 Protein Information
Protein Expression
Protein Purification
4.2.2. NS4A
4.2.3. DSLS Binding Test
4.2.4. Binding and Competition Assay by Fluorescence Anisotropy
4.2.5. Enzyme Inhibition Assay
4.3. Molecular Modeling
4.3.1. Hardware and Software
4.3.2. Protein and Ligand Preparation
4.3.3. Molecular Dynamics
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Hepatitis Report. 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 21 January 2020).
- Friedrich, M.J. Third millennium challenge: Hepatitis C. JAMA 1999, 282, 221–222. [Google Scholar] [PubMed]
- Dickson, R.C. Clinical manifestations of hepatitis C. Clin. Liver Dis. 1997, 1, 569–585. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- North, C.S.; Hong, B.A.; Adewuyi, S.A.; Pollio, D.E.; Jain, M.K.; Devereaux, R.; Quartey, N.A.; Ashitey, S.; Lee, W.M.; Lisker-Melman, M. Hepatitis C treatment and SVR: The gap between clinical trials and real-world treatment aspirations. Gen. Hosp. Psychiatry 2013, 35, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Panel, A.-I.H.G. Hepatitis C Guidance 2018 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Clin. Infect. Dis. 2018, 67, 1477–1492. [Google Scholar] [CrossRef] [Green Version]
- Chahine, E.B.; Kelley, D.; Childs-Kean, L.M. Sofosbuvir/Velpatasvir/Voxilaprevir: A Pan-Genotypic Direct-Acting Antiviral Combination for Hepatitis C. Ann. Pharmacother. 2018, 52, 352–363. [Google Scholar] [CrossRef]
- Chhatwal, J.; Chen, Q.; Aggarwal, R. Estimation of Hepatitis C Disease Burden and Budget Impact of Treatment Using Health Economic Modeling. Infect. Dis. Clin. N. Am. 2018, 32, 461–480. [Google Scholar] [CrossRef]
- Cuypers, L.; Libin, P.; Schrooten, Y.; Theys, K.; Di Maio, V.C.; Cento, V.; Lunar, M.M.; Nevens, F.; Poljak, M.; Ceccherini-Silberstein, F. Exploring resistance pathways for first-generation NS3/4A protease inhibitors boceprevir and telaprevir using Bayesian network learning. Infect. Genet. Evol. 2017, 53, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Li, D.K.; Chung, R.T. Overview of Direct-Acting Antiviral Drugs and Drug Resistance of Hepatitis C Virus. Methods Mol. Biol. 2019, 1911, 3–32. [Google Scholar] [CrossRef]
- Hellard, M.E.; Chou, R.; Easterbrook, P. WHO Guidelines on Testing for Hepatitis B and C–Meeting Targets for Testing; BioMed Central: London, UK, 2017. [Google Scholar]
- Bartenschlager, R.; Baumert, T.F.; Bukh, J.; Houghton, M.; Lemon, S.M.; Lindenbach, B.D.; Lohmann, V.; Moradpour, D.; Pietschmann, T.; Rice, C.M.; et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: Considerations for scientists and funding agencies. Virus Res. 2018, 248, 53–62. [Google Scholar] [CrossRef]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: Roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Microbiol. 2008, 6, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; O’malley, E.T. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Gwack, Y.; Han, J.H.; Choe, J. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem. Biophys. Res. Commun. 1995, 215, 160–166. [Google Scholar] [CrossRef]
- Failla, C.; Tomei, L.; De Francesco, R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 1994, 68, 3753–3760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Pragai, B.M.; Grakoui, A.; Xu, J.; Rice, C.M. Hepatitis C virus NS3 serine proteinase: Trans-cleavage requirements and processing kinetics. J. Virol. 1994, 68, 8147–8157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, K.; Lange, C.M.; Gouttenoire, J.; Meylan, E.; Brass, V.; Penin, F.; Moradpour, D. Nonstructural protein 3–4A: The Swiss army knife of hepatitis C virus. J. Viral Hepat. 2011, 18, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- De Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [Green Version]
- El-Araby, M.E.; Omar, A.M.; El-Faky, M.A.; Soror, S.H.; Khayat, M.T.; Asfour, H.Z.; Bamane, F. Imidazole derivatives as HCV NS3/4A protease. Preprints 2019. [Google Scholar] [CrossRef]
- Yan, Y.; Li, Y.; Munshi, S.; Sardana, V.; Cole, J.L.; Sardana, M.; Steinkuehler, C.; Tomei, L.; De Francesco, R.; Kuo, L.C.; et al. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: A 2.2 Å resolution structure in a hexagonal crystal form. Protein Sci. 1998, 7, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Thomson, J.A.; Rice, C.M. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3–NS4A serine proteinase complex in vivo and in vitro. J. Virol. 1995, 69, 4373–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butkiewicz, N.J.; Wendel, M.; Zhang, R.; Jubin, R.; Pichardo, J.; Smith, E.B.; Hart, A.M.; Ingram, R.; Durkin, J.; Mui, P.W.; et al. Enhancement of hepatitis C virus NS3 proteinase activity by association with NS4A-specific synthetic peptides: Identification of sequence and critical residues of NS4A for the cofactor activity. Virology 1996, 225, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Yamaji, K.; Masuho, Y.; Yokota, T.; Inoue, H.; Sudo, K.; Satoh, S.; Shimotohno, K. Identification of the sequence on NS4A required for enhanced cleavage of the NS5A/5B site by hepatitis C virus NS3 protease. J. Virol. 1996, 70, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomei, L.; Failla, C.; Vitale, R.L.; Bianchi, E.; De Francesco, R. A central hydrophobic domain of the hepatitis C virus NS4A protein is necessary and sufficient for the activation of the NS3 protease. J. Gen. Virol. 1996, 77, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- El-Araby, M.E.; Omar, A.M.; El-Faky, M.A.; Soror, S.H.; Khayat, M.T.; Asfour, H.Z.; Bamane, F.H. Peptide Inhibitors of HCV NS3/4A Protease Comprising Non-Proteinogenic Amino Residues. U.S. Patent Application 16/371,610, 1 April 2019. [Google Scholar]
- El-Araby, M.E.; Omar, A.M.; Soror, S.H.; Arold, S.T.; Khayat, M.T.; Asfour, H.Z.; Bamane, F.; El-Faky, M.A. Synthetic bulky NS4A peptide variants bind to and inhibit HCV NS3 protease. J. Adv. Res. 2020. [Google Scholar] [CrossRef]
- Joyce, M.; Williams, M.; Hindsgaul, O.; Tyrrell, D. Inhibitors of Hepatitis C Virus Protease. U.S. Patent US20030176689A1, 18 September 2003. [Google Scholar]
- De Francesco, R.; Pessi, A.; Steinkühler, C. Mechanisms of hepatitis C virus NS3 proteinase inhibitors. J. Viral Hepat. 1999, 6, 23–30. [Google Scholar] [CrossRef]
- Hamad, H.A.; Thurston, J.; Teague, T.; Ackad, E.; Yousef, M.S. The NS4A cofactor dependent enhancement of HCV NS3 protease activity correlates with a 4D geometrical measure of the catalytic triad region. PLoS ONE 2016, 11, e0168002. [Google Scholar] [CrossRef]
- Jiang, Y.; Andrews, S.W.; Condroski, K.R.; Buckman, B.; Serebryany, V.; Wenglowsky, S.; Kennedy, A.L.; Madduru, M.R.; Wang, B.; Lyon, M.; et al. Discovery of danoprevir (ITMN-191/R7227), a highly selective and potent inhibitor of hepatitis C virus (HCV) NS3/4A protease. J. Med. Chem. 2014, 57, 1753–1769. [Google Scholar] [CrossRef]
- Soumana, D.I.; Kurt Yilmaz, N.; Ali, A.; Prachanronarong, K.L.; Schiffer, C.A. Molecular and Dynamic Mechanism Underlying Drug Resistance in Genotype 3 Hepatitis C NS3/4A Protease. J. Am. Chem. Soc. 2016, 138, 11850–11859. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.G.; Zipfel, S.; Ramey, K.; Vivian, R.; Schrier, A.; Karki, K.K.; Katana, A.; Kato, D.; Kobayashi, T.; Martinez, R.; et al. Discovery of the pan-genotypic hepatitis C virus NS3/4A protease inhibitor voxilaprevir (GS-9857): A component of Vosevi®. Bioorg. Med. Chem. Lett. 2019, 29, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- Aloup, J.-C.; Audiau, F.; Barreau, M.; Damour, D.; Genevois-Borella, A.; Jimonet, P.; Mignani, S.; Ribeill, Y. Preparation of Imidazo [1,2-a]Indeno [1,2-e]Pyrazine-2-Carboxylates as AMPA and NMDA Receptor Antagonists. U.S. Patent WO9602544A1, 1 February 1996. [Google Scholar]
- Aloup, J.-C.; Bouquerel, J.; Damour, D.; Hardy, J.-C.; Mignani, S. 2-Substituted 5H,10H-Imidazo[1,2-a]Indeno[1,2-e]Pyrazin-4-Ones, Useful as AMPA and NMDA Receptor Antagonists, Their Preparation, and Drugs Containing Them. U.S. Patent WO9725326A1, 17 July 1997. [Google Scholar]
- Bauer, L.; Nambury, C.N.V.; Bell, C.L. Studies in the chemistry of 3- and 5-isoxazolones. Tetrahedron 1964, 20, 165–171. [Google Scholar] [CrossRef]
- Sarabia, F.; Sánchez-Ruiz, A.; Chammaa, S. Stereoselective synthesis of E-64 and related cysteine proteases inhibitors from 2,3-epoxyamides. Bioorg. Med. Chem. 2005, 13, 1691–1705. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Niesen, F.H.; Allali-Hassani, A.; Fedorov, O.Y.; Finerty, P.J., Jr.; Wasney, G.A.; Yeung, R.; Arrowsmith, C.; Ball, L.J.; Berglund, H.; et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA 2006, 103, 15835–15840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senisterra, G.A.; Markin, E.; Yamazaki, K.; Hui, R.; Vedadi, M.; Awrey, D.E. Screening for ligands using a generic and high-throughput light-scattering-based assay. J. Biomol. Screen. 2006, 11, 940–948. [Google Scholar] [CrossRef] [Green Version]
- Shahul Hameed, U.; Haider, I.; Jamil, M.; Kountche, B.A.; Guo, X.; Zarban, R.A.; Kim, D.; Al-Babili, S.; Arold, S.T. Structural basis for specific inhibition of the highly sensitive ShHTL7 receptor. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Hamill, P.; Jean, F. Enzymatic Characterization of Membrane-Associated Hepatitis C Virus NS3-4A Heterocomplex Serine Protease Activity Expressed in Human Cells. Biochemistry 2005, 44, 6586–6596. [Google Scholar] [CrossRef]
- Vora, J.; Patel, S.; Athar, M.; Sinha, S.; Chhabria, M.T.; Jha, P.C.; Shrivastava, N. Pharmacophore modeling, molecular docking and molecular dynamics simulation for screening and identifying anti-dengue phytocompounds. J. Biomol. Struct. Dyn. 2019, 1–15. [Google Scholar] [CrossRef]
- Pérez-Benito, L.; Keränen, H.; van Vlijmen, H.; Tresadern, G. Predicting Binding Free Energies of PDE2 Inhibitors. The Difficulties of Protein Conformation. Sci. Rep. 2018, 8, 4883. [Google Scholar] [CrossRef] [Green Version]
- Cummings, M.D.; Lindberg, J.; Lin, T.I.; de Kock, H.; Lenz, O.; Lilja, E.; Fellander, S.; Baraznenok, V.; Nystrom, S.; Nilsson, M.; et al. Induced-fit binding of the macrocyclic noncovalent inhibitor TMC435 to its HCV NS3/NS4A protease target. Angew. Chem. Int. Ed. Engl. 2010, 49, 1652–1655. [Google Scholar] [CrossRef]
- Bennett, F.; Huang, Y.; Hendrata, S.; Lovey, R.; Bogen, S.L.; Pan, W.; Guo, Z.; Prongay, A.; Chen, K.X.; Arasappan, A.; et al. The introduction of P4 substituted 1-methylcyclohexyl groups into Boceprevir: A change in direction in the search for a second generation HCV NS3 protease inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 2617–2621. [Google Scholar] [CrossRef] [PubMed]
- Prongay, A.J.; Guo, Z.; Yao, N.; Pichardo, J.; Fischmann, T.; Strickland, C.; Myers, J., Jr.; Weber, P.C.; Beyer, B.M.; Ingram, R.; et al. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3- [2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]- 6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem. 2007, 50, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, A.; Allam, N.; Elsharkway, A.; El Kassas, M.; Waked, I. Hepatitis C infection in Egypt: Prevalence, impact and management strategies. Hepatic Med. Evid. Res. 2017, 9, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamoud, Y.A.; Riome, S.; Abu-Raddad, L.J. Epidemiology of hepatitis C virus in the Arabian Gulf countries: Systematic review and meta-analysis of prevalence. Int. J. Infect. Dis. 2016, 46, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massariol, M.-J.; Zhao, S.; Marquis, M.; Thibeault, D.; White, P.W. Protease and helicase activities of hepatitis C virus genotype 4, 5, and 6 NS3–NS4A proteins. Biochem. Biophys. Res. Commun. 2010, 391, 692–697. [Google Scholar] [CrossRef]
- Bawazir, A.; AlGusheri, F.; Jradi, H.; AlBalwi, M.; Abdel-Gader, A.-G. Hepatitis C virus genotypes in Saudi Arabia: A future prediction and laboratory profile. Virol. J. 2017, 14, 208. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Competition IC50 ± SEM (µM) |
|---|---|
| MOC-11 | 17.3 ± 1.22 |
| MOC-23 | 4.70 ± 0.327 |
| MOC-24 | 1.91 ± 0.119 |
| MOC-26 | >100 |
| MOC-27 | >100 |
| MOC-28 | >100 |
| MOC-29 | >100 |
| MOC-30 | 28.5 ± 8.55 |
| MOC-31 | 12.1 ± 1.69 |
| MOC-32 | 7.74 ± 0.240 |
| MOC-33 | 40.3 ± 24.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omar, A.M.; Elfaky, M.A.; Arold, S.T.; Soror, S.H.; Khayat, M.T.; Asfour, H.Z.; Bamane, F.H.; El-Araby, M.E. 1H-Imidazole-2,5-Dicarboxamides as NS4A Peptidomimetics: Identification of a New Approach to Inhibit HCV-NS3 Protease. Biomolecules 2020, 10, 479. https://doi.org/10.3390/biom10030479
Omar AM, Elfaky MA, Arold ST, Soror SH, Khayat MT, Asfour HZ, Bamane FH, El-Araby ME. 1H-Imidazole-2,5-Dicarboxamides as NS4A Peptidomimetics: Identification of a New Approach to Inhibit HCV-NS3 Protease. Biomolecules. 2020; 10(3):479. https://doi.org/10.3390/biom10030479
Chicago/Turabian StyleOmar, Abdelsattar M., Mahmoud A. Elfaky, Stefan T. Arold, Sameh H. Soror, Maan T. Khayat, Hani Z. Asfour, Faida H. Bamane, and Moustafa E. El-Araby. 2020. "1H-Imidazole-2,5-Dicarboxamides as NS4A Peptidomimetics: Identification of a New Approach to Inhibit HCV-NS3 Protease" Biomolecules 10, no. 3: 479. https://doi.org/10.3390/biom10030479
APA StyleOmar, A. M., Elfaky, M. A., Arold, S. T., Soror, S. H., Khayat, M. T., Asfour, H. Z., Bamane, F. H., & El-Araby, M. E. (2020). 1H-Imidazole-2,5-Dicarboxamides as NS4A Peptidomimetics: Identification of a New Approach to Inhibit HCV-NS3 Protease. Biomolecules, 10(3), 479. https://doi.org/10.3390/biom10030479