BET Inhibitors Synergize with Carfilzomib to Induce Cell Death in Cancer Cells via Impairing Nrf1 Transcriptional Activity and Exacerbating the Unfolded Protein Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. MTT Assays for Measuring Cell Viability
2.3. Determination of CI
2.4. Quantitative Reverse Transcription PCR
2.5. Proteasome Activity Recovery Assay
2.6. Luciferase Assays
2.7. Immunoblot Analysis
2.8. RNA Sequencing and Analysis
2.9. Statistical Analysis
3. Results
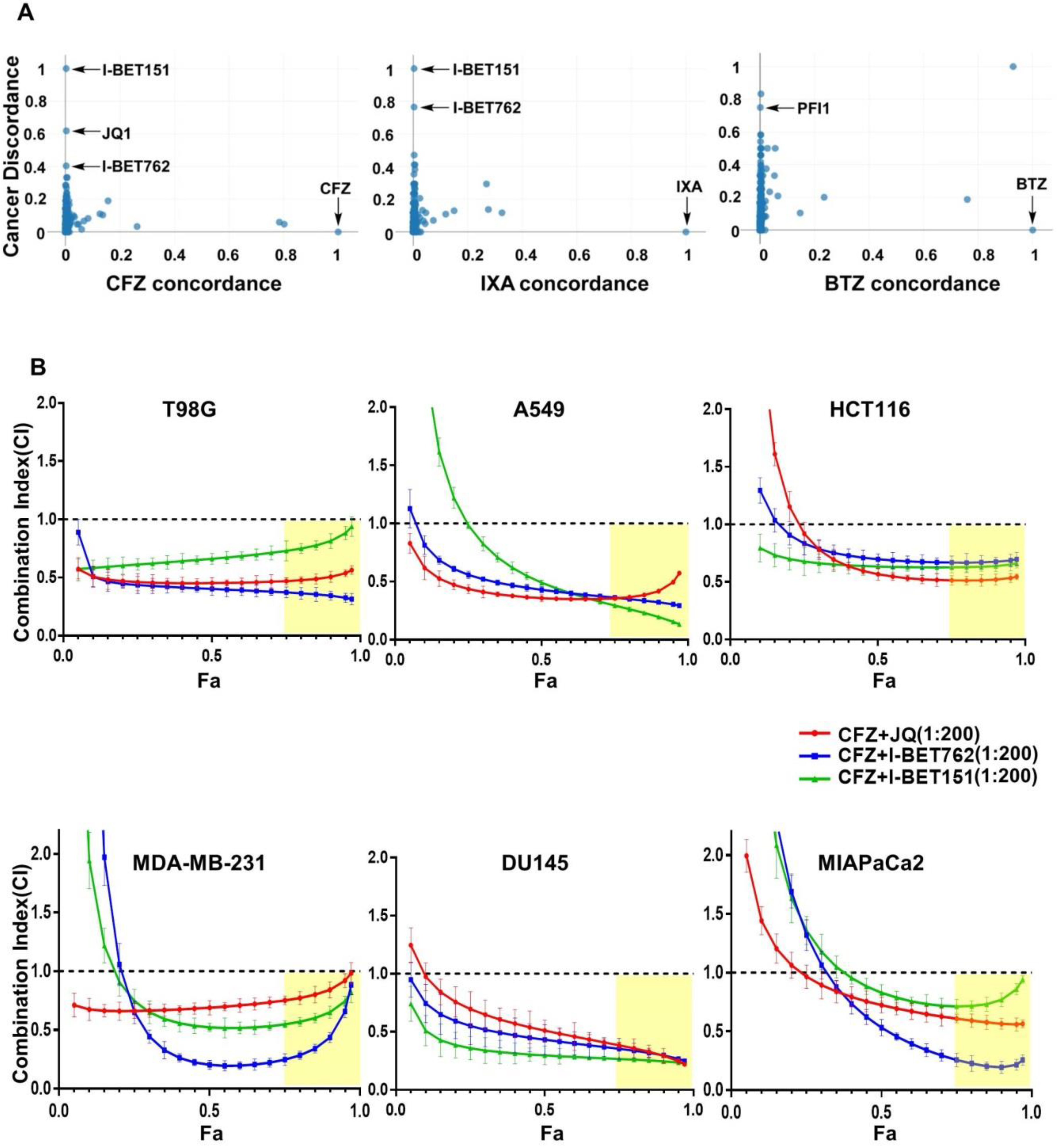
3.1. Identification of BET Inhibitors as Synergizers of Proteasome Inhibitor-Induced Cancer Cell Death
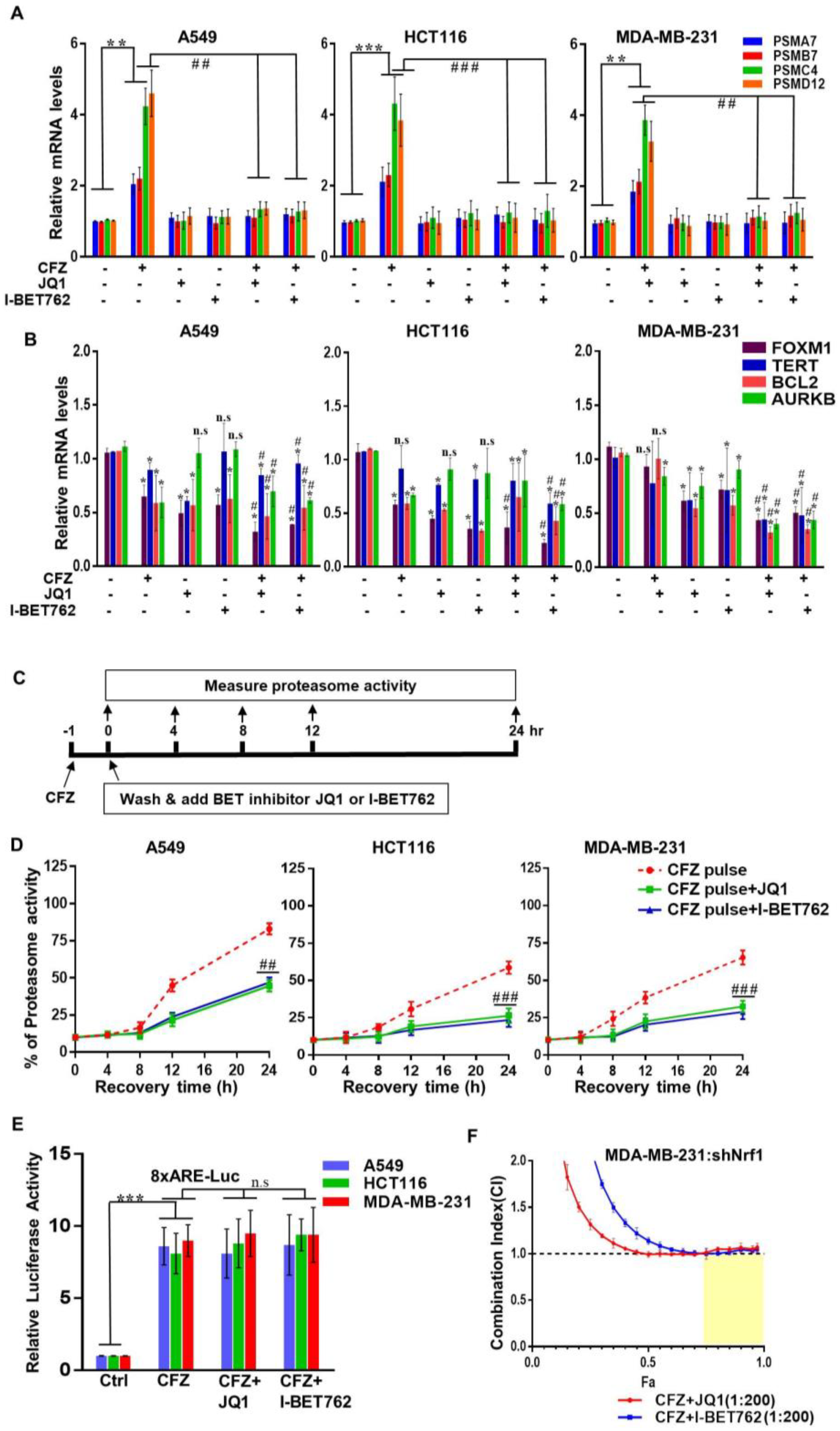
3.2. BET Inhibitors Attenuate CFZ-Mediated Nrf1-Dependent Proteasome Bounce-Back Response
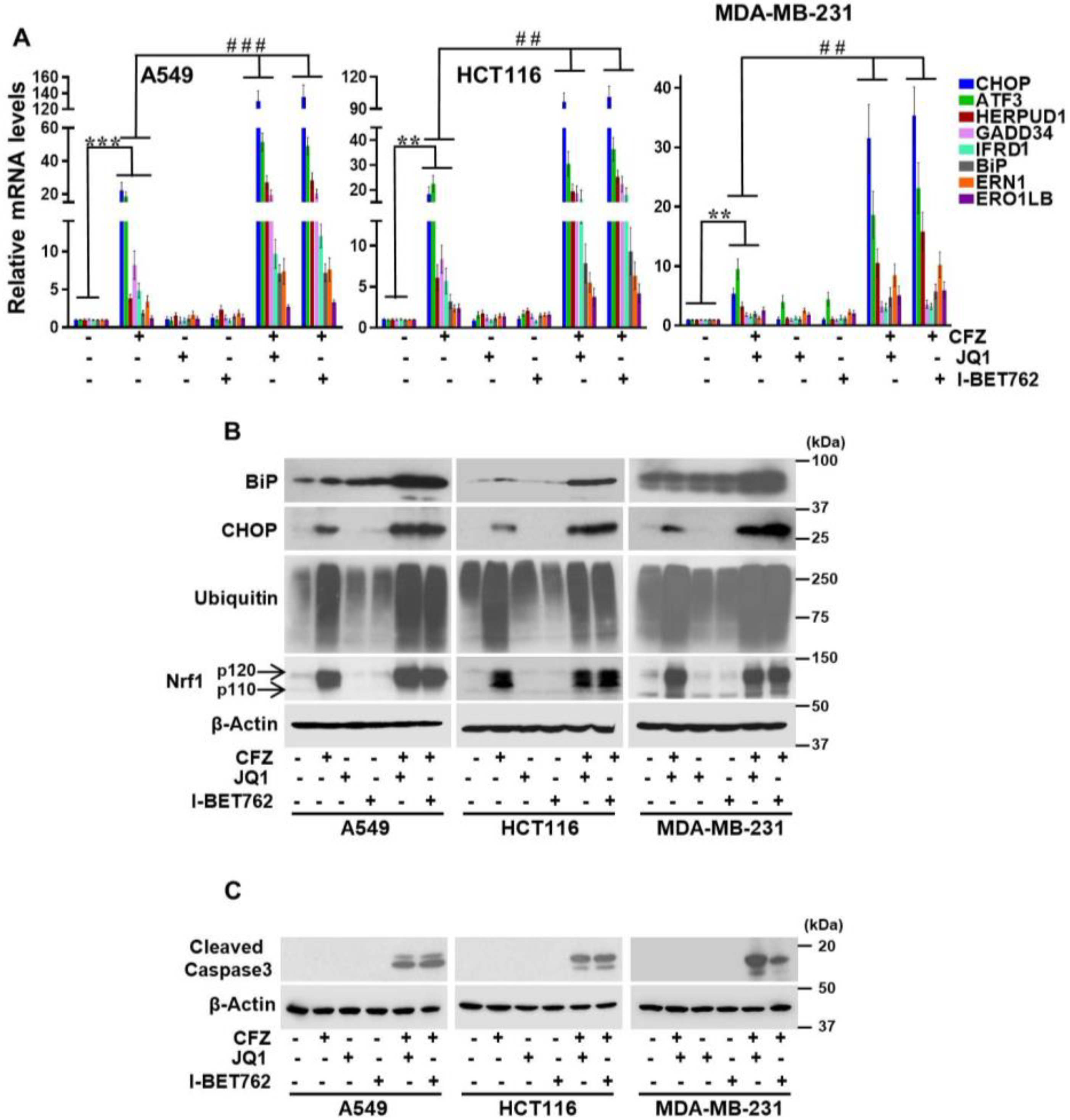
3.3. BET Inhibitors Exacerbate CFZ-Mediated Unfolded Protein Response (UPR)
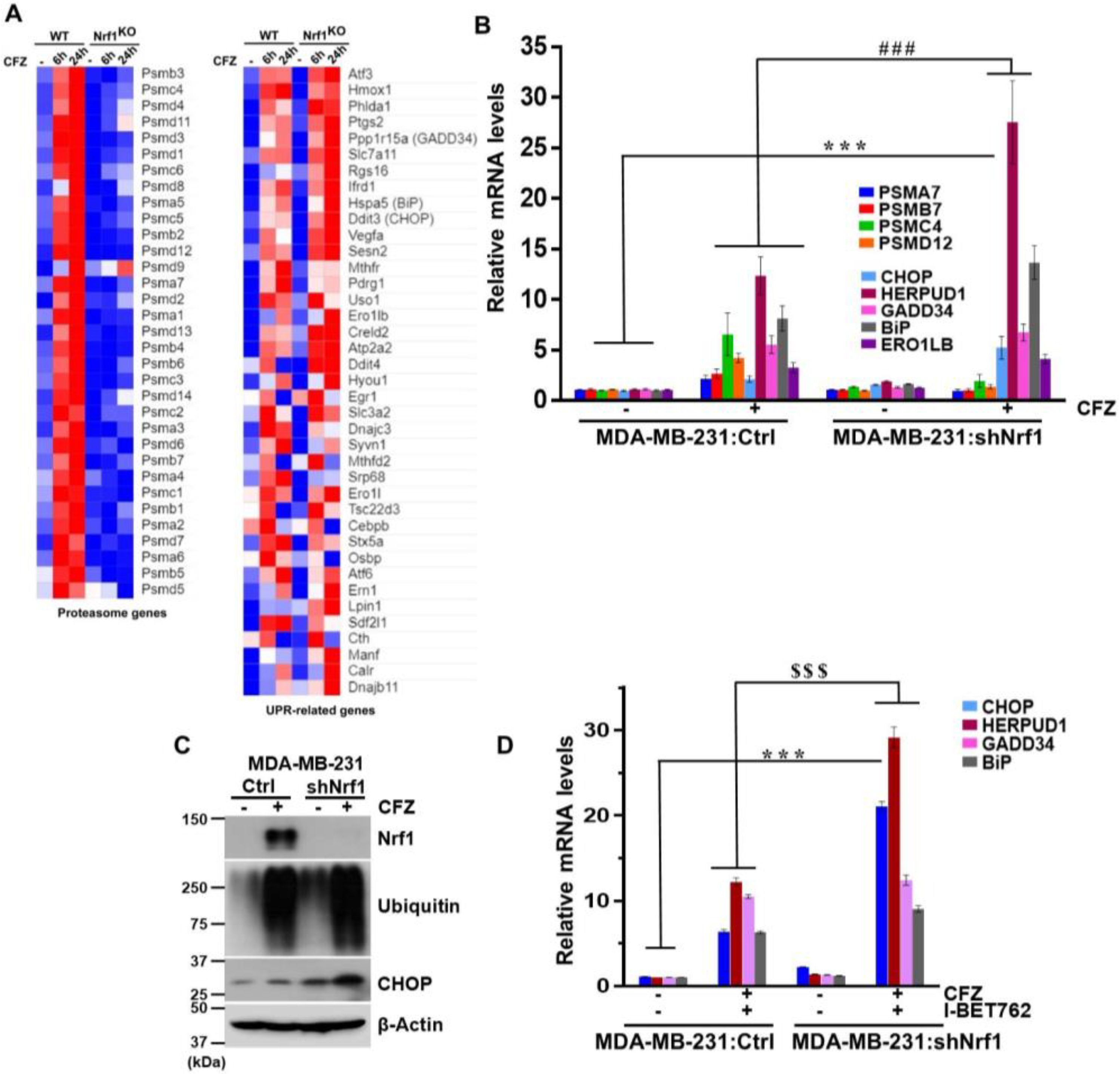
3.4. Depletion of Nrf1 Exacerbates CFZ-Mediated UPR
4. Discussion
Supplementary Materials
Data Availability
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Tomko, R.J., Jr.; Hochstrasser, M. Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, P.; Adler, J.; Myers, N.; Biran, A.; Reuven, N.; Shaul, Y. Oncogenic addiction to high 26S proteasome level. Cell Death Dis. 2018, 9, 773. [Google Scholar] [CrossRef] [Green Version]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef]
- Holkova, B.; Grant, S. Proteasome inhibitors in mantle cell lymphoma. Best Pract. Res. Clin. Haematol. 2012, 25, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.P.; Lonial, S.; Boise, L.H. When Cancer Fights Back: Multiple Myeloma, Proteasome Inhibition, and the Heat-Shock Response. Mol. Cancer Res. 2015, 13, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Gu, Y.; Ta, L.; Wang, K.; Xu, Z. Induction of autophagy by the MG132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF7 cells. Mol. Med. Rep. 2016, 13, 796–804. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Dunner, K., Jr.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014, 3, e01856. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Vangala, J.R.; Radhakrishnan, S.K. Nrf1-mediated transcriptional regulation of the proteasome requires a functional TIP60 complex. J. Biol. Chem. 2019, 294, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Vangala, J.R.; Sotzny, F.; Kruger, E.; Deshaies, R.J.; Radhakrishnan, S.K. Nrf1 can be processed and activated in a proteasome-independent manner. Curr. Biol. 2016, 26, R834–R835. [Google Scholar] [CrossRef] [Green Version]
- Williamson, M.J.; Silva, M.D.; Terkelsen, J.; Robertson, R.; Yu, L.; Xia, C.; Hatsis, P.; Bannerman, B.; Babcock, T.; Cao, Y.; et al. The relationship among tumor architecture, pharmacokinetics, pharmacodynamics, and efficacy of bortezomib in mouse xenograft models. Mol. Cancer Ther. 2009, 8, 3234–3243. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.C.; Kalebic, T.; Infante, J.R.; Siu, L.L.; Sullivan, D.; Vlahovic, G.; Kauh, J.S.; Gao, F.; Berger, A.J.; Tirrell, S.; et al. Phase 1 study of ixazomib, an investigational proteasome inhibitor, in advanced non-hematologic malignancies. Investig. New Drugs 2015, 33, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Weyburne, E.S.; Wilkins, O.M.; Sha, Z.; Williams, D.A.; Pletnev, A.A.; de Bruin, G.; Overkleeft, H.S.; Goldberg, A.L.; Cole, M.D.; Kisselev, A.F. Inhibition of the Proteasome beta2 Site Sensitizes Triple-Negative Breast Cancer Cells to beta5 Inhibitors and Suppresses Nrf1 Activation. Cell Chem. Biol. 2017, 24, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yu, Y.; Wang, Z.; Wang, H.; Bieerkehazhi, S.; Zhao, Y.; Suzuk, L.; Zhang, H. Second-generation proteasome inhibitor carfilzomib enhances doxorubicin-induced cytotoxicity and apoptosis in breast cancer cells. Oncotarget 2016, 7, 73697–73710. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, Y.; Jiang, Z.; Cao, W.M.; Wang, Z.; Dou, J.; Zhao, Y.; Cui, Y.; Zhang, H. Next-generation proteasome inhibitor MLN9708 sensitizes breast cancer cells to doxorubicin-induced apoptosis. Sci. Rep. 2016, 6, 26456. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.; Dunner, K., Jr.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef] [Green Version]
- Chang, I.; Wang, C.Y. Inhibition of HDAC6 Protein Enhances Bortezomib-induced Apoptosis in Head and Neck Squamous Cell Carcinoma (HNSCC) by Reducing Autophagy. J. Biol. Chem. 2016, 291, 18199–18209. [Google Scholar] [CrossRef] [Green Version]
- Iovine, B.; Iannella, M.L.; Nocella, F.; Pricolo, M.R.; Bevilacqua, M.A. Carnosine inhibits KRAS-mediated HCT116 proliferation by affecting ATP and ROS production. Cancer Lett. 2012, 315, 122–128. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, X.; Chen, L.; Weng, S.; Xia, Y.; Ye, Y.; Li, K.; Liao, Z.; Chen, P.; Alsamman, K.; et al. Regulation of the Expression of DAPK1 by SUMO Pathway. Biomolecules 2019, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Federici, G.; Gilardini Montani, M.S.; Crispini, A.; Cirone, M.; D’Orazi, G. Interplay between Endoplasmic Reticulum (ER) Stress and Autophagy Induces Mutant p53H273 Degradation. Biomolecules 2020, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; Gartel, A.L. The PPAR-gamma agonist pioglitazone post-transcriptionally induces p21 in PC3 prostate cancer but not in other cell lines. Cell Cycle 2005, 4, 582–584. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Rekha, K.; Rao, R.R.; Pandey, R.; Prasad, K.R.; Babu, K.S.; Vangala, J.R.; Kalivendi, S.V.; Rao, J.M. Two new sesquiterpenoids from the rhizomes of Nardostachys jatamansi. J. Asian Nat. Prod. Res. 2013, 15, 111–116. [Google Scholar] [CrossRef]
- Yadav, J.S.; Boyapelly, K.; Alugubelli, S.R.; Pabbaraja, S.; Vangala, J.R.; Kalivendi, S.V. Stereoselective total synthesis of (+)-oploxyne A, (-)-oploxyne B, and their C-10 epimers and structure revision of natural oploxyne B. J. Org. Chem. 2011, 76, 2568–2576. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Vangala, J.R.; Dudem, S.; Jain, N.; Kalivendi, S.V. Regulation of PSMB5 protein and beta subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (STAT3): Potential role in bortezomib-mediated anticancer therapy. J. Biol. Chem. 2014, 289, 12612–12622. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Stathias, V.; Jermakowicz, A.M.; Maloof, M.E.; Forlin, M.; Walters, W.; Suter, R.K.; Durante, M.A.; Williams, S.L.; Harbour, J.W.; Volmar, C.H.; et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat. Commun. 2018, 9, 5315. [Google Scholar] [CrossRef]
- Blum, A.; Wang, P.; Zenklusen, J.C. SnapShot: TCGA-Analyzed Tumors. Cell 2018, 173, 530. [Google Scholar] [CrossRef]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Morita, T.; Kim, M.; Iemura, S.; Natsume, T.; Yamamoto, M.; Kobayashi, A. Dual regulation of the transcriptional activity of Nrf1 by beta-TrCP- and Hrd1-dependent degradation mechanisms. Mol. Cell. Biol. 2011, 31, 4500–4512. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pena, J.; Gyorffy, B.; Amir, E.; Pandiella, A.; Ocana, A. Epigenetic modulation of FOXM1-gene interacting network by BET inhibitors in breast cancer. Breast Cancer Res. Treat. 2018, 172, 725–732. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Northrop, A.; Vangala, J.R.; Feygin, A.; Radhakrishnan, S.K. Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 327. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5, e18357. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 2016, 5, e17721. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell. Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vangala, J.R.; Potluri, A.; Radhakrishnan, S.K. BET Inhibitors Synergize with Carfilzomib to Induce Cell Death in Cancer Cells via Impairing Nrf1 Transcriptional Activity and Exacerbating the Unfolded Protein Response. Biomolecules 2020, 10, 501. https://doi.org/10.3390/biom10040501
Vangala JR, Potluri A, Radhakrishnan SK. BET Inhibitors Synergize with Carfilzomib to Induce Cell Death in Cancer Cells via Impairing Nrf1 Transcriptional Activity and Exacerbating the Unfolded Protein Response. Biomolecules. 2020; 10(4):501. https://doi.org/10.3390/biom10040501
Chicago/Turabian StyleVangala, Janakiram R., Ajay Potluri, and Senthil K. Radhakrishnan. 2020. "BET Inhibitors Synergize with Carfilzomib to Induce Cell Death in Cancer Cells via Impairing Nrf1 Transcriptional Activity and Exacerbating the Unfolded Protein Response" Biomolecules 10, no. 4: 501. https://doi.org/10.3390/biom10040501
APA StyleVangala, J. R., Potluri, A., & Radhakrishnan, S. K. (2020). BET Inhibitors Synergize with Carfilzomib to Induce Cell Death in Cancer Cells via Impairing Nrf1 Transcriptional Activity and Exacerbating the Unfolded Protein Response. Biomolecules, 10(4), 501. https://doi.org/10.3390/biom10040501