A Novel NAD-RNA Decapping Pathway Discovered by Synthetic Light-Up NAD-RNAs

 , ,
, ,
Abstract
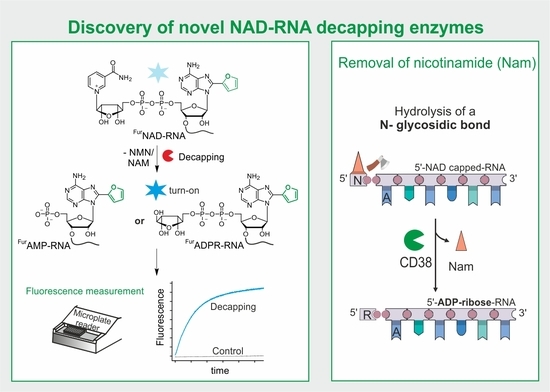
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Synthesis of 8-(furan-2-yl)-NAD
2.3. Synthesis of Oligonucleotides
2.4. Synthesis of 5’-FurNAD-RNAs
2.5. HPLC Analysis and Purification
2.6. Mass Spectrometric Analysis
2.7. In Vitro Transcription (IVT)
2.8. Cloning of Different Nudix Hydrolases
2.9. Overexpression and Affinity Purification of Different Nudix Hydrolases
2.10. Fluorescence Spectroscopy Using a Sensitive Cuvette Spectrofluorometer
2.11. Fluorescence Time Course Measurements Using a Fluorescence Microplate Reader
2.12. Radioactive Validation of CD38 Activity
3. Results
3.1. Chemo-Enzymatic Synthesis of FurNAD and FurNAD-RNA
3.2. Fluorescence Properties of FurNAD-RNAs
3.3. Screening for Novel NAD-RNA Decapping Enzymes
3.4. Determination of Kinetic Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Topisirovic, I.; Svitkin, Y.V.; Sonenberg, N.; Shatkin, A.J. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip. Rev. RNA 2011, 2, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Kowtoniuk, W.E.; Agarwal, I.; Shen, Y.; Liu, D.R. LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat. Chem. Biol. 2009, 5, 879–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahova, H.; Winz, M.L.; Höfer, K.; Nübel, G.; Jäschke, A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 2015, 519, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Jäschke, A.; Höfer, K.; Nübel, G.; Frindert, J. Cap-like structures in bacterial RNA and epitranscriptomic modification. Curr. Opin. Microbiol. 2016, 30, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Winz, M.L.; Cahová, H.; Nübel, G.; Frindert, J.; Höfer, K.; Jäschke, A. Capture and sequencing of NAD-capped RNA sequences with NAD captureSeq. Nat. Protoc. 2017, 12, 122–149. [Google Scholar] [CrossRef] [PubMed]
- Luciano, D.J.; Belasco, J.G. NAD in RNA: Unconventional headgear. Trends Biochem. Sci 2015, 40, 245–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marbaniang, C.N.; Vogel, J. Emerging roles of RNA modifications in bacteria. Curr. Opin. Microbiol. 2016, 30, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Frindert, J.; Zhang, Y.; Nubel, G.; Kahloon, M.; Kolmar, L.; Hotz-Wagenblatt, A.; Burhenne, J.; Haefeli, W.E.; Jäschke, A. Identification, Biosynthesis, and Decapping of NAD-Capped RNAs in B. subtilis. Cell Rep. 2018, 24, 1890–1901.e8. [Google Scholar] [CrossRef] [Green Version]
- Morales-Filloy, H.G.; Zhang, Y.; Nübel, G.; George, S.E.; Korn, N.; Wolz, C.; Jäschke, A. The 5’-NAD cap of RNAIII modulates toxin production in Staphylococcus aureus isolates. J. Bacteriol. 2019. [Google Scholar] [CrossRef]
- Jiao, X.; Doamekpor, S.K.; Bird, J.G.; Nickels, B.E.; Tong, L.; Hart, R.P.; Kiledjian, M. 5’ End Nicotinamide Adenine Dinucleotide Cap in Human Cells Promotes RNA Decay through DXO-Mediated deNADding. Cell 2017, 168, 1015–1027.e10. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.W.; Matheny, T.; Mizoue, L.S.; Rao, B.S.; Muhlrad, D.; Parker, R. Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, S.; Zhao, Y.; You, C.; Le, B.; Gong, Z.; Mo, B.; Xia, Y.; Chen, X. NAD(+)-capped RNAs are widespread in the Arabidopsis transcriptome and can probably be translated. Proc. Natl. Acad. Sci. USA 2019, 116, 12094–12102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhong, H.; Zhang, S.; Shao, X.; Ni, M.; Cai, Z.; Chen, X.; Xia, Y. NAD tagSeq reveals that NAD(+)-capped RNAs are mostly produced from a large number of protein-coding genes in Arabidopsis. Proc. Natl. Acad. Sci. USA 2019, 116, 12072–12077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, J.G.; Zhang, Y.; Tian, Y.; Panova, N.; Barvik, I.; Greene, L.; Liu, M.; Buckley, B.; Krasny, L.; Lee, J.K.; et al. The mechanism of RNA 5’ capping with NAD+, NADH and desphospho-CoA. Nature 2016, 533, 444–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vvedenskaya, I.O.; Bird, J.G.; Zhang, Y.; Zhang, Y.; Jiao, X.; Barvik, I.; Krasny, L.; Kiledjian, M.; Taylor, D.M.; Ebright, R.H.; et al. CapZyme-Seq Comprehensively Defines Promoter-Sequence Determinants for RNA 5’ Capping with NAD(). Mol. Cell 2018, 70, 553–564.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julius, C.; Yuzenkova, Y. Noncanonical RNA-capping: Discovery, mechanism, and physiological role debate. Wiley Interdiscip. Rev. RNA 2019, 10, e1512. [Google Scholar] [CrossRef] [PubMed]
- Dunckley, T.; Parker, R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J. 1999, 18, 5411–5422. [Google Scholar] [CrossRef]
- Jiao, X.; Chang, J.H.; Kilic, T.; Tong, L.; Kiledjian, M. A mammalian pre-mRNA 5’ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol. Cell 2013, 50, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Picard-Jean, F.; Brand, C.; Tremblay-Letourneau, M.; Allaire, A.; Beaudoin, M.C.; Boudreault, S.; Duval, C.; Rainville-Sirois, J.; Robert, F.; Pelletier, J.; et al. 2’-O-methylation of the mRNA cap protects RNAs from decapping and degradation by DXO. PLoS ONE 2018, 13, e0193804. [Google Scholar]
- Grudzien-Nogalska, E.; Kiledjian, M. New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kwasnik, A.; Wang, V.Y.; Krzyszton, M.; Gozdek, A.; Zakrzewska-Placzek, M.; Stepniak, K.; Poznanski, J.; Tong, L.; Kufel, J. Arabidopsis DXO1 links RNA turnover and chloroplast function independently of its enzymatic activity. Nucleic Acids Res. 2019, 47, 4751–4764. [Google Scholar] [CrossRef] [PubMed]
- Song, M.G.; Bail, S.; Kiledjian, M. Multiple Nudix family proteins possess mRNA decapping activity. RNA 2013, 19, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celesnik, H.; Deana, A.; Belasco, J.G. Pablo Analysis of RNA: 5 ’-Phosphorylation state and 5’-end mapping. Methods Enzymol. 2008, 447, 83–98. [Google Scholar] [PubMed]
- Nübel, G.; Sorgenfrei, F.A.; Jäschke, A. Boronate affinity electrophoresis for the purification and analysis of cofactor-modified RNAs. Methods 2017, 117, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Jiao, X.; Welch, S.; Kiledjian, M. Analysis of mRNA decapping. Methods Enzymol. 2008, 448, 3–21. [Google Scholar] [PubMed]
- Kellner, S.; Burhenne, J.; Helm, M. Detection of RNA modifications. RNA Biol. 2010, 7, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, M.; Reichle, V.F.; Kellner, S. Observing the fate of tRNA and its modifications by nucleic acid isotope labeling mass spectrometry: NAIL-MS. RNA Biol. 2017, 14, 1260–1268. [Google Scholar] [CrossRef] [Green Version]
- Pesnot, T.; Kempter, J.; Schemies, J.; Pergolizzi, G.; Uciechowska, U.; Rumpf, T.; Sippl, W.; Jung, M.; Wagner, G.K. Two-step synthesis of novel, bioactive derivatives of the ubiquitous cofactor nicotinamide adenine dinucleotide (NAD). J. Med. Chem. 2011, 54, 3492–3499. [Google Scholar] [CrossRef]
- Pergolizzi, G.; Butt, J.N.; Bowater, R.P.; Wagner, G.K. A novel fluorescent probe for NAD-consuming enzymes. Chem. Commun. (Camb.) 2011, 47, 12655–12657. [Google Scholar] [CrossRef] [Green Version]
- Höfer, K.; Abele, F.; Schlotthauer, J.; Jäschke, A. Synthesis of 5’-NAD-Capped RNA. Bioconjug. Chem. 2016, 27, 874–877. [Google Scholar] [CrossRef]
- Wagner, G.K.; Pesnot, T.; Field, R.A. A survey of chemical methods for sugar-nucleotide synthesis. Nat. Prod. Rep. 2009, 26, 1172–1194. [Google Scholar] [CrossRef]
- Höfer, K.; Li, S.; Abele, F.; Frindert, J.; Schlotthauer, J.; Grawenhoff, J.; Du, J.; Patel, D.J.; Jäschke, A. Structure and function of the bacterial decapping enzyme NudC. Nat. Chem. Biol. 2016, 12, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Greco, N.J.; Tor, Y. Furan decorated nucleoside analogues as fluorescent probes: Synthesis, photophysical evaluation, and site-specific incorporation. Tetrahedron 2007, 63, 3515–3527. [Google Scholar] [CrossRef] [PubMed]
- Huang, F. Efficient incorporation of CoA, NAD and FAD into RNA by in vitro transcription. Nucleic Acids Res. 2003, 31, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.Q.; Wang, G.C.; Coleman, T.; Li, N. Synthesis of adenosine derivatives as transcription initiators and preparation of 5 ’ fluorescein- and biotin-labeled RNA through one-step in vitro transcription. RNA 2003, 9, 1562–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, A.; Krause, A.; Jäschke, A. A modified dinucleotide for site-specific RNA-labelling by transcription priming and click chemistry. Chem. Commun. (Camb) 2014, 50, 1313–1316. [Google Scholar] [CrossRef]
- Frick, D.N.; Bessman, M.J. Cloning, Purification, and Properties of a Novel Nadh Pyrophosphatase–Evidence for a Nucleotide Pyrophosphatase Catalytic Domain in Mutt-Like Enzymes. J. Biol. Chem. 1995, 270, 1529–1534. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C. Enzymatic functions and structures of CD38 and homologs. Chem. Immunol. 2000, 75, 39–59. [Google Scholar]
- O’Handley, S.F.; Frick, D.N.; Dunn, C.A.; Bessman, M.J. Orf186 represents a new member of the Nudix hydrolases, active on adenosine(5’)triphospho(5’)adenosine, ADP-ribose, and NADH. J. Biol. Chem. 1998, 273, 3192–3197. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Aarhus, R. ADP-Ribosyl Cyclase–an Enzyme That Cyclizes NAD+ into a Calcium-Mobilizing Metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Gasmi, L.; Cartwright, J.L.; McLennan, A.G. Cloning, expression and characterization of YSA1H, a human adenosine 5 ’-diphosphosugar pyrophosphatase possessing a MutT motif. Biochem. J. 1999, 344, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Qi, S.L.; Unger, M.; Hou, Y.N.; Deng, Q.W.; Liu, J.; Lam, C.M.C.; Wang, X.W.; Xin, D.; Zhang, P.; et al. Immuno-targeting the multifunctional CD38 using nanobody. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.J.; Zhu, W.J.; Wang, X.W.; Zhang, L.H.; Lee, H.C. Determinants of the membrane orientation of a calcium signaling enzyme CD38. Biochim. Biophys. Acta 2015, 1853, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Arimori, T.; Tamaoki, H.; Nakamura, T.; Kamiya, H.; Ikemizu, S.; Takagi, Y.; Ishibashi, T.; Harashima, H.; Sekiguchi, M.; Yamagata, Y. Diverse substrate recognition and hydrolysis mechanisms of human NUDT5. Nucleic Acids Res. 2011, 39, 8972–8983. [Google Scholar] [CrossRef] [Green Version]
- Höfer, K.; Jäschke, A. Epitranscriptomics: RNA Modifications in Bacteria and Archaea. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Graeff, R.M.; Walseth, T.F.; Fryxell, K.; Branton, W.D.; Lee, H.C. Enzymatic synthesis and characterizations of cyclic GDP-ribose. A procedure for distinguishing enzymes with ADP-ribosyl cyclase activity. J. Biol. Chem. 1994, 269, 30260–30267. [Google Scholar]
- de Oliveira, G.C.; Kanamori, K.S.; Auxiliadora-Martins, M.; Chini, C.C.S.; Chini, E.N. Measuring CD38 Hydrolase and Cyclase Activities: 1,N(6)-Ethenonicotinamide Adenine Dinucleotide (epsilon-NAD) and Nicotinamide Guanine Dinucleotide (NGD) Fluorescence-based Methods. Bio. Protoc. 2018, 8. [Google Scholar] [CrossRef]
- Graeff, R.; Munshi, C.; Aarhus, R.; Johns, M.; Lee, H.C. A single residue at the active site of CD38 determines its NAD cyclizing and hydrolyzing activities. J. Biol. Chem. 2001, 276, 12169–12173. [Google Scholar] [CrossRef] [Green Version]
- Kramer, S.; McLennan, A.G. The complex enzymology of mRNA decapping: Enzymes of four classes cleave pyrophosphate bonds. Wiley Interdiscip. Rev. RNA 2019, 10, e1511. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.L.; Liu, Y.X.; Wang, Q.; Guan, Z.Y.; Wang, J.; Liu, J.; Zou, T.T.; Yin, P. Structural basis of prokaryotic NAD-RNA decapping by NudC. Cell Res. 2016, 26, 1062–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AbdelRaheim, S.R.; Cartwright, J.L.; Gasmi, L.; McLennan, A.G. The NADH diphosphatase encoded by the Saccharomyces cerevisiae NYP1 nudix hydrolase gene is located in peroxisomes. Arch. Biochem. Biophys. 2001, 388, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Jiao, X.; Chiba, K.; Oh, C.; Martin, C.E.; Kiledjian, M.; Tong, L. Dxo1 is a new type of eukaryotic enzyme with both decapping and 5’-3’ exoribonuclease activity. Nat. Struct. Mol. Biol. 2012, 19, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sydorskyy, Y.; Dilworth, D.J.; Yi, E.C.; Goodlett, D.R.; Wozniak, R.W.; Aitchison, J.D. Intersection of the Kap123p-mediated nuclear import and ribosome export pathways. Mol. Cell. Biol. 2003, 23, 2042–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abele, F.; Höfer, K.; Bernhard, P.; Grawenhoff, J.; Seidel, M.; Krause, A.; Kopf, S.; Schröter, M.; Jäschke, A. A Novel NAD-RNA Decapping Pathway Discovered by Synthetic Light-Up NAD-RNAs. Biomolecules 2020, 10, 513. https://doi.org/10.3390/biom10040513
Abele F, Höfer K, Bernhard P, Grawenhoff J, Seidel M, Krause A, Kopf S, Schröter M, Jäschke A. A Novel NAD-RNA Decapping Pathway Discovered by Synthetic Light-Up NAD-RNAs. Biomolecules. 2020; 10(4):513. https://doi.org/10.3390/biom10040513
Chicago/Turabian StyleAbele, Florian, Katharina Höfer, Patrick Bernhard, Julia Grawenhoff, Maximilian Seidel, André Krause, Sara Kopf, Martin Schröter, and Andres Jäschke. 2020. "A Novel NAD-RNA Decapping Pathway Discovered by Synthetic Light-Up NAD-RNAs" Biomolecules 10, no. 4: 513. https://doi.org/10.3390/biom10040513
APA StyleAbele, F., Höfer, K., Bernhard, P., Grawenhoff, J., Seidel, M., Krause, A., Kopf, S., Schröter, M., & Jäschke, A. (2020). A Novel NAD-RNA Decapping Pathway Discovered by Synthetic Light-Up NAD-RNAs. Biomolecules, 10(4), 513. https://doi.org/10.3390/biom10040513