Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase


Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Protein Crystallisation
2.3. Diffraction Data Collection and Structure Determination
2.4. Single Turnover Fluorescence Assay
2.5. Analysis of Protein Stability
2.6. Analysis of the Protein Oligomeric State
2.7. FRET Measurements
3. Results
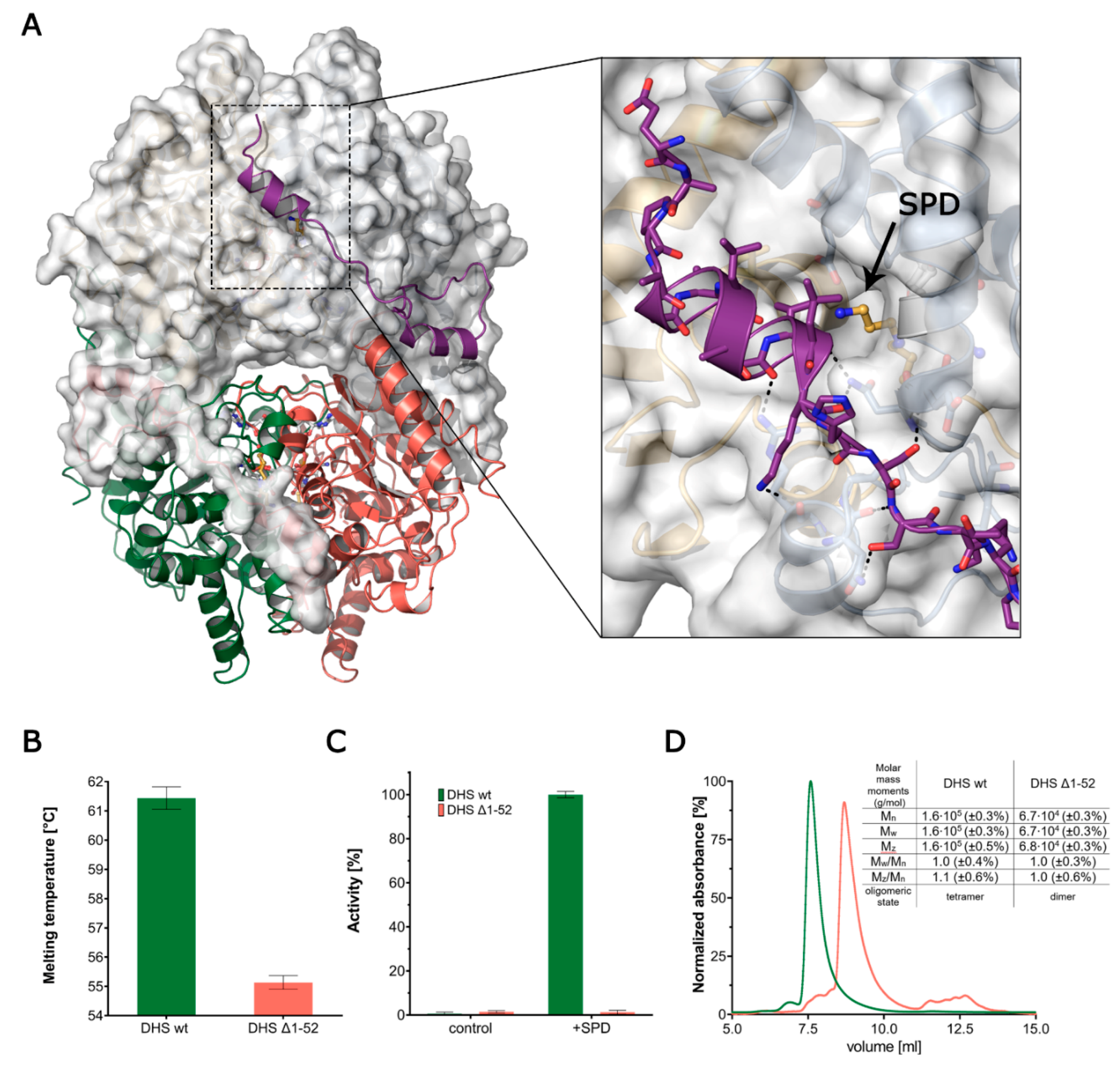
3.1. DHS Forms a Functional Tetramer
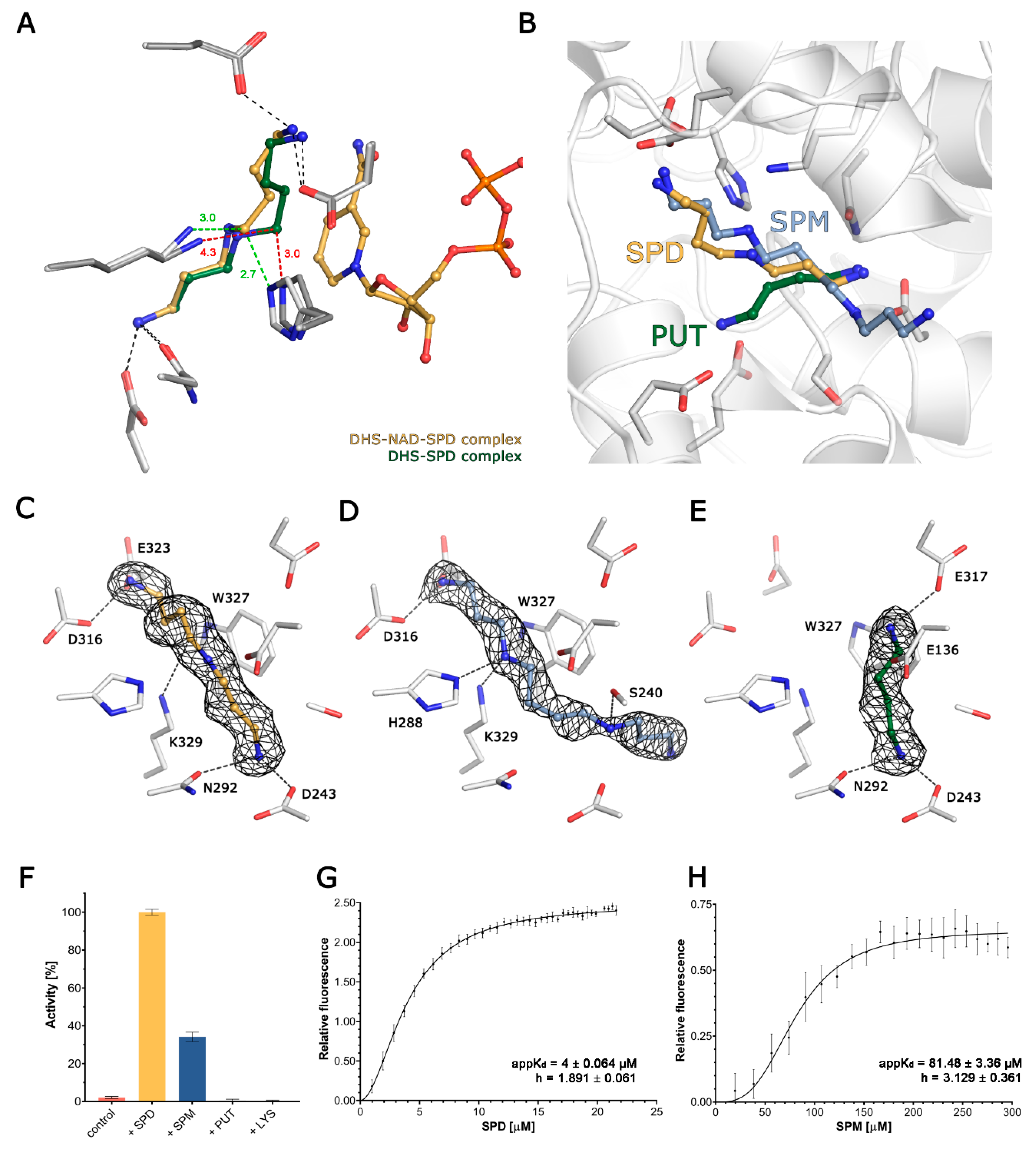
3.2. Crystal Structures of Binary DHS-Spermidine and Ternary DHS-Spermidine-NAD Complexes
3.3. DHS Binds Spermine and Putrescine
3.4. Structural Comparison of Polyamine Binding by DHS
3.5. Spermidine and Spermine Cooperatively Bind DHS Active Sites
3.6. The Ball-and-Chain Motif Is Crucial for DHS Activity, Stability and Oligomerisation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, H.L.; Park, M.H.; Folk, J.E. Posttranslational formation of hypusine in a single major protein occurs generally in growing cells and is associated with activation of lymphocyte growth. Cell 1982, 29, 791–797. [Google Scholar] [CrossRef]
- Park, M.H. The Essential Role of Hypusine in Eukaryotic Translation Initiation. J. Biol. Chem. 1989, 264, 18531–18535. [Google Scholar] [PubMed]
- Park, M.H.; Wolff, E.C.; Smit-McBride, Z.; Hershey, J.W.B.; Folk, J.E. Comparison of the activities of variant forms of eIF-4D: The requirement for hypusine or deoxyhypusine. J. Biol. Chem. 1991, 266, 7988–7994. [Google Scholar]
- Wolff, E.C.; Kang, K.R.; Kim, Y.S.; Park, M.H. Posttranslational synthesis of hypusine: Evolutionary progression and specificity of the hypusine modification. Amino Acids 2007, 71, 233–236. [Google Scholar]
- Park, M.H.; Wolff, E.C. Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 2018, 293, 18710–18718. [Google Scholar] [CrossRef] [Green Version]
- Saini, P.; Eyler, D.E.; Green, R.; Dever, T.E. Hypusine-containing Protein eIF5A Promotes Translation Elongation. Nature 2011, 459, 118–121. [Google Scholar] [CrossRef] [Green Version]
- Pelechano, V.; Alepuz, P. EIF5A facilitates translation termination globally and promotes the elongation of many non polyproline-specific tripeptide sequences. Nucleic Acids Res. 2017, 45, 7326–7338. [Google Scholar] [CrossRef] [Green Version]
- Lassak, J.; Wilson, D.N.; Jung, K. Stall no more at polyproline stretches with the translation elongation factors EF-P and IF-5A. Mol. Microbiol. 2016, 99, 219–235. [Google Scholar] [CrossRef] [Green Version]
- Mandal, A.; Mandal, S.; Park, M.H. Genome-wide analyses and functional classification of proline repeat-rich proteins: Potential role of eIF5A in eukaryotic evolution. PLoS ONE 2014, 9, e111800. [Google Scholar] [CrossRef] [Green Version]
- Schnier, J.; Schwelberger, H.G.; Smit-McBride, Z.; Kang, H.A.; Hershey, J.W. Translation initiation factor 5A and its hypusine modification are essential for cell viability in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 3105–3114. [Google Scholar] [CrossRef]
- Pällmann, N.; Braig, M.; Sievert, H.; Preukschas, M.; Hermans-Borgmeyer, I.; Schweizer, M.; Nagel, C.H.; Neumann, M.; Wild, P.; Haralambieva, E.; et al. Biological relevance and therapeutic potential of the hypusine modification system. J. Biol. Chem. 2015, 290, 18343–18360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Wolff, E.C.; Folk, J.E. Is hypusine essential for eukaryotic cell proliferation? Trends Biochem. Sci. 1993, 18, 475–479. [Google Scholar] [CrossRef]
- Oliverio, S.; Corazzari, M.; Sestito, C.; Piredda, L.; Ippolito, G.; Piacentini, M. The spermidine analogue GC7 (N1-guanyl-1,7-diamineoheptane) induces autophagy through a mechanism not involving the hypusination of eIF5A. Amino Acids 2014, 46, 2767–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, A. Translational control of eIF5A in various diseases. Amino Acids 2012, 42, 679–684. [Google Scholar] [CrossRef]
- Hoque, M.; Hanauske-Abel, H.M.; Palumbo, P.; Saxena, D.; D’Alliessi Gandolfi, D.; Park, M.H.; Pe’ery, T.; Mathews, M.B. Inhibition of HIV-1 gene expression by Ciclopirox and Deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. Retrovirology 2009, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, S.; Cleveland, J.L. Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino Acids 2016, 48, 2353–2362. [Google Scholar] [CrossRef] [Green Version]
- Maier, B.; Nadler, J.L.; Raghavendra, G.; Maier, B.; Ogihara, T.; Trace, A.P.; Tersey, S.A.; Robbins, R.D.; Chakrabarti, S.K.; Dondero, R.S.; et al. The unique hypusine modification of eIF5A promotes islet β cell inflammation and dysfunction in mice. J. Clin. Investig. 2010, 12, 2156–2170. [Google Scholar] [CrossRef] [Green Version]
- Caraglia, M.; Park, M.H.; Wolff, E.C.; Marra, M.; Abbruzzese, A. EIF5A isoforms and cancer: Two brothers for two functions? Amino Acids 2013, 44, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Park, I.; Hong, B.-S.; Nedyalkova, L.; Tempel, W.; Park, H.-W. Crystal structure of human eIF5A1: Insight into functional similarity of human eIF5A1 and eIF5A2. Proteins 2009, 75, 1040–1045. [Google Scholar] [CrossRef]
- Clement, P.M.J.; Henderson, C.A.; Jenkins, Z.A.; Smit-McBride, Z.; Wolff, E.C.; Hershey, J.W.B.; Park, M.H.; Johansson, H.E. Identification and characterization of eukaryotic initiation factor 5A-2. Eur. J. Biochem. 2003, 270, 4254–4263. [Google Scholar] [CrossRef]
- Scuoppo, C.; Miething, C.; Lindqvist, L.; Reyes, J.; Ruse, C.; Appelmann, I.; Yoon, S.; Krasnitz, A.; Teruya-Feldstein, J.; Pappin, D.; et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature 2012, 487, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.B.; Kang, W.M.; Yu, J.C.; Liu, Y.Q.; Ma, Z.Q.; Zhou, L.; Cui, Q.-C.; Zhou, W.-X. Overexpression of eukaryotic translation initiation factor 5A2 (EIF5A2) correlates with cell aggressiveness and poor survival in gastric cancer. PLoS ONE 2015, 10, e0119229. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, H.; Chen, Y.; Wei, J.; Chen, Z.; Feng, Z.; Huang, Y.; Chen, W.; Luo, J.; Fang, Y. Eukaryotic translation initiation factor 5A2 is highly expressed in prostate cancer and predicts poor prognosis. Exp. Ther. Med. 2019, 17, 3741–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantuano, E.; Trettel, F.; Olsen, A.S.; Lennon, G.; Frontali, M.; Jodice, C. Localization and genomic structure of human deoxyhypusine synthase gene on chromosome 19p13.2-distal 19p13.1. Gene 1998, 215, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Wolff, E.C.; Wolff, J.; Park, M.H. Deoxyhypusine synthase generates and uses bound NADH in a transient hydride transfer mechanism. J. Biol. Chem. 2000, 275, 9170–9177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, E.C.; Park, M.H.; Folk, J.E. Cleavage of spermidine as the first step in deoxyhypusine synthesis. The role of NAD+. J. Biol. Chem. 1990, 265, 4793–4799. [Google Scholar]
- Wolff, E.C.; Folk, J.E.; Park, M.H. Enzyme-substrate intermediate formation at lysine 329 of human deoxyhypusine synthase. J. Biol. Chem. 1997, 272, 15865–15871. [Google Scholar] [CrossRef] [Green Version]
- Joe, Y.A.; Wolff, E.C.; Lee, Y.B.; Park, M.H. Enzyme-Substrate Intermediate at a Specific Lysine Residue Is Required for Deoxyhypusine Synthesis. J. Biol. Chem. 2002, 272, 32679–32685. [Google Scholar] [CrossRef] [Green Version]
- Myung, H.P.; Wolff, E.C.; Young, B.L.; Folk, J.E. Antiproliferative effects of inhibitors of deoxyhypusine synthase. Inhibition of growth of Chinese hamster ovary cells by guanyl diamines. J. Biol. Chem. 1994, 269, 27827–27832. [Google Scholar]
- Templin, A.T.; Maier, B.; Nishiki, Y.; Tersey, S.A.; Mirmira, R.G. Deoxyhypusine synthase haploinsufficiency attenuates acute cytokine signaling. Cell Cycle 2011, 10, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Ganapathi, M.; Padgett, L.R.; Yamada, K.; Devinsky, O.; Willaert, R.; Person, R.; Au, P.-Y.B.; Tagoe, J.; McDonald, M.; Karlowicz, D.; et al. Recessive Rare Variants in Deoxyhypusine Synthase, an Enzyme Involved in the Synthesis of Hypusine, Are Associated with a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 104, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kim, H.K.; Park, H.E.; Park, M.H.; Joe, Y.A. Effect of N1-guanyl-1,7-diaminoheptane, an inhibitor of deoxyhypusine synthase, on endothelial cell growth, differentiation and apoptosis. Mol. Cell. Biochem. 2002, 237, 69–76. [Google Scholar] [CrossRef]
- Lee, S.K.; Lee, J.; Lee, S.I.; Bae, W.J.; Lee, Y.M.; Park, J.S.; Lee, S.-K.; Park, S.-J.; Min, S.-K.; Kim, E.-C. N1-guanyl-1,7,-diamineoheptane, an inhibitor of deoxyhypusine synthase, suppresses differentiation and induces apoptosis via mitochondrial and AMPK pathways in immortalized and malignant human oral keratinocytes. J. Oral. Pathol. Med. 2009, 38, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.R.; Geerts, D.; Mooney, M.; El-Khawaja, R.; Koster, J.; Bachmann, A.S. Synergistic drug combination GC7/DFMO suppresses hypusine/spermidine-dependent eIF5A activation and induces apoptotic cell death in neuroblastoma. Biochem. J. 2018, 475, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.U.; Ignatenko, N.A.; Yamauchi, P.; Stringer, D.E.; Levenson, C.; Shannon, P.; Shannon, P.; Perrin, S.; Gerner, E.W. Inhibition of human ornithine decarboxylase activity by enantiomers of difluoromethylornithine. Biochem. J. 2003, 375, 465–470. [Google Scholar]
- Umland, T.C.; Wolff, E.C.; Park, M.H.; Davies, D.R. A new crystal structure of deoxyhypusine synthase reveals the configuration of the active enzyme and of an enzyme·NAD·inhibitor ternary complex. J. Biol. Chem. 2004, 279, 28697–28705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, D.-I.; Wolff, E.C.; Park, M.H.; Davies, D.R. Crystal structure of the NAD complex of human deoxyhypusine synthase: An enzyme with a ball-and-chain mechanism for blocking the active site. Structure 2004, 6, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Gorrec, F. Protein crystallization screens developed at the MRC Laboratory of Molecular Biology. Drug Discov. Today 2016, 21, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Mueller, U.; Darowski, N.; Fuchs, M.R.; Förster, R.; Hellmig, M.; Paithankar, K.S.; Pühringer, S.; Steffien, M.; Zocher, G.; Weiss, M.S. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron. Radiat. 2012, 19, 442–449. [Google Scholar] [CrossRef]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. Xdsapp2.0. J. Appl. Crystallogr. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanador, G.A.; Tomchick, D.R.; Phillips, M.A. Trypanosomatid Deoxyhypusine Synthase Activity Is Dependent on Shared Active-Site Complementation between Pseudoenzyme Paralogs. Structure 2018, 26, 1499–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhard, L.; Mayerhofer, H.; Geerlof, A.; Mueller-Dieckmann, J.; Weiss, M.S. Optimization of protein buffer cocktails using Thermofluor. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Park, M.H. Human deoxyhypusine synthase: Interrelationship between binding of NAD and substrates. Biochem. J. 2000, 352, 851–857. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Hoon, L.C.; Um, P.Y.; Hee, P.M. Structure–function studies of human deoxyhypusine synthase: Identification of amino acid residues critical for the binding of spermidine and NAD. Biochem. J. 2001, 355, 841–849. [Google Scholar]
- Igarashi, K.; Kashiwagi, K. The functional role of polyamines in eukaryotic cells. Int. J. Biochem. Cell Biol. 2019, 107, 104–115. [Google Scholar] [CrossRef]
- Laube, G.; Bernstein, H.G. Agmatine: Multifunctional arginine metabolite and magic bullet in clinical neuroscience? Biochem. J. 2017, 474, 2619–2640. [Google Scholar] [CrossRef] [PubMed]
- Šečkute, J.; McCloskey, D.E.; Thomas, H.J.; Secrist, J.A.; Pegg, A.E.; Ealick, S.E. Binding and inhibition of human spermidine synthase by decarboxylated S-adenosylhomocysteine. Protein Sci. 2011, 20, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Wolff, E.C.; Folk, J.E.; Park, M.H. Reversal of the deoxyhypusine synthesis reaction: Generation of spermidine or homospermidine from deoxyhypusine by deoxyhypusine synthase. J. Biol. Chem. 2003, 278, 32683–32691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.J.; Wolff, E.C.; Myung, H.P. Cloning and expression of human deoxyhypusine synthase cDNA. Structure- function studies with the recombinant enzyme and mutant proteins. J. Biol. Chem. 1995, 270, 22386–22392. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| DHS Structure | apo | NAD | NAD-SPD | SPD | SPM | PUT |
|---|---|---|---|---|---|---|
| PDB ID | 6XXH | 6XXI | 6XXJ | 6XXK | 6XXL | 6XXM |
| Wavelength (Å) | 0.9184 | 0.9184 | 0.9184 | 0.9184 | 0.9184 | 0.9184 |
| Resolution range (Å) | 46.22–1.52 (1.57–1.52) * | 46.09–1.68 (1.74–1.68) | 46.24–1.41 (1.46–1.41) | 46.19–1.65 (1.71–1.65) | 46.08–1.69 (1.75–1.69) | 46.12–1.67 (1.73–1.67) |
| Space group | P 32 2 1 | P 32 2 1 | P 32 2 1 | P 32 2 1 | P 32 2 1 | P 32 2 1 |
| Unit cell (Å,°) | 104.97 104.97 161.03 90.0 90.0 120.0 | 104.82 104.82 160.50 90.0 90.0 120.0 | 105.49 105.49 160.83 90.0 90.0 120.0 | 104.98 104.98 160.90 90.0 90.0 120.0 | 104.90 104.90 160.43 90.0 90.0 120.0 | 104.73 104.73 160.67 90.0 90.0 120.0 |
| Total reflections | 1592643 (257143) | 979651 (159039) | 1946524 (308384) | 1371934 (220304) | 2283917 (365779) | 1191506 (182975) |
| Unique reflections | 157636 (15594) | 116336 (18588) | 198531 (19596) | 123673 (12190) | 114734 (11265) | 118316 (11679) |
| Multiplicity | 10.10 | 8.42 | 9.80 | 11.09 | 19.90 | 6.51 |
| Completeness (%) | 99.92 (99.88) | 99.61 (98.77) | 99.82 (99.48) | 99.80 (99.05) | 99.71 (98.40) | 99.81 (99.45) |
| Mean I/sigma(I) | 13.09 (0.76) | 8.88 (0.92) | 13.34 (0.76) | 12.80 (0.94) | 17.69 (0.75) | 14.28 (0.95) |
| Wilson B-factor | 22.41 | 26.10 | 20.06 | 26.06 | 31.73 | 25.73 |
| R-merge (%) | 10.6 (304.6) | 12.6 (202.1) | 8.8 (243.7) | 11.0 (235.2) | 10.5 (433.3) | 10.6 (258.6) |
| R-meas (%) | 11.1 (320.7) | 13.5 (215.3) | 9.4 (257.3) | 11.6 (246.5) | 10.8 (444.4) | 11.2 (272.9) |
| R-sym (%) | 10.6 (323.2) | 12.5 (220.7) | 9.1 (308.5) | 11.3 (282.9) | 10.8 (506.8) | 10.8 (278.1) |
| CC1/2 (%) | 99.9 (43.4) | 99.6 (73.7) | 99.9 (54.8) | 99.9 (67.2) | 100.0 (58.9) | 99.9 (49.6) |
| Reflections used in refinement | 157555 (15581) | 116220 (11408) | 198411 (19572) | 123538 (12161) | 114606 (11231) | 118271 (11666) |
| Reflections used for R-free | 2099 (208) | 2097 (205) | 2096 (207) | 2098 (207) | 2099 (206) | 2098 (206) |
| R-work (%) | 15.42 (34.35) | 15.39 (34.91) | 14.25 (34.82) | 15.53 (36.48) | 16.17 (39.78) | 16.05 (33.51) |
| R-free (%) | 17.62 (34.86) | 16.62 (36.76) | 15.98 (36.52) | 16.22 (38.77) | 17.93 (45.02) | 17.46 (36.44) |
| Number of non-hydrogen atoms | 6095 | 5916 | 6443 | 6013 | 6057 | 6186 |
| macromolecules | 5320 | 5317 | 5635 | 5439 | 5477 | 5596 |
| ligands | 22 | 243 | 215 | 169 | 107 | 20 |
| solvent | 753 | 356 | 593 | 405 | 473 | 570 |
| Protein residues | 667 | 679 | 692 | 691 | 675 | 692 |
| RMS (bonds) | 0.010 | 0.021 | 0.011 | 0.018 | 0.013 | 0.010 |
| RMS (angles) | 1.10 | 1.77 | 1.17 | 1.43 | 1.31 | 1.10 |
| Ramachandran favored (%) | 98.02 | 98.63 | 98.97 | 97.94 | 98.05 | 98.67 |
| Ramachandran allowed (%) | 1.98 | 1.37 | 1.03 | 2.06 | 1.95 | 1.33 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Rotamer outliers (%) | 0.35 | 0.35 | 0.33 | 0.00 | 0.67 | 0.33 |
| Clashscore | 3.00 | 3.43 | 5.47 | 5.28 | 6.78 | 3.02 |
| Average B-factor | 34.78 | 39.05 | 29.52 | 36.93 | 47.86 | 38.77 |
| macromolecules | 32.88 | 37.87 | 27.97 | 35.76 | 46.59 | 37.79 |
| ligands | 51.62 | 54.97 | 44.86 | 55.55 | 69.85 | 55.71 |
| solvent | 47.71 | 45.85 | 38.63 | 44.94 | 57.59 | 47.81 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wątor, E.; Wilk, P.; Grudnik, P. Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase. Biomolecules 2020, 10, 522. https://doi.org/10.3390/biom10040522
Wątor E, Wilk P, Grudnik P. Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase. Biomolecules. 2020; 10(4):522. https://doi.org/10.3390/biom10040522
Chicago/Turabian StyleWątor, Elżbieta, Piotr Wilk, and Przemysław Grudnik. 2020. "Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase" Biomolecules 10, no. 4: 522. https://doi.org/10.3390/biom10040522
APA StyleWątor, E., Wilk, P., & Grudnik, P. (2020). Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase. Biomolecules, 10(4), 522. https://doi.org/10.3390/biom10040522