P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases


Abstract
:1. Introduction
2. Materials and Methods
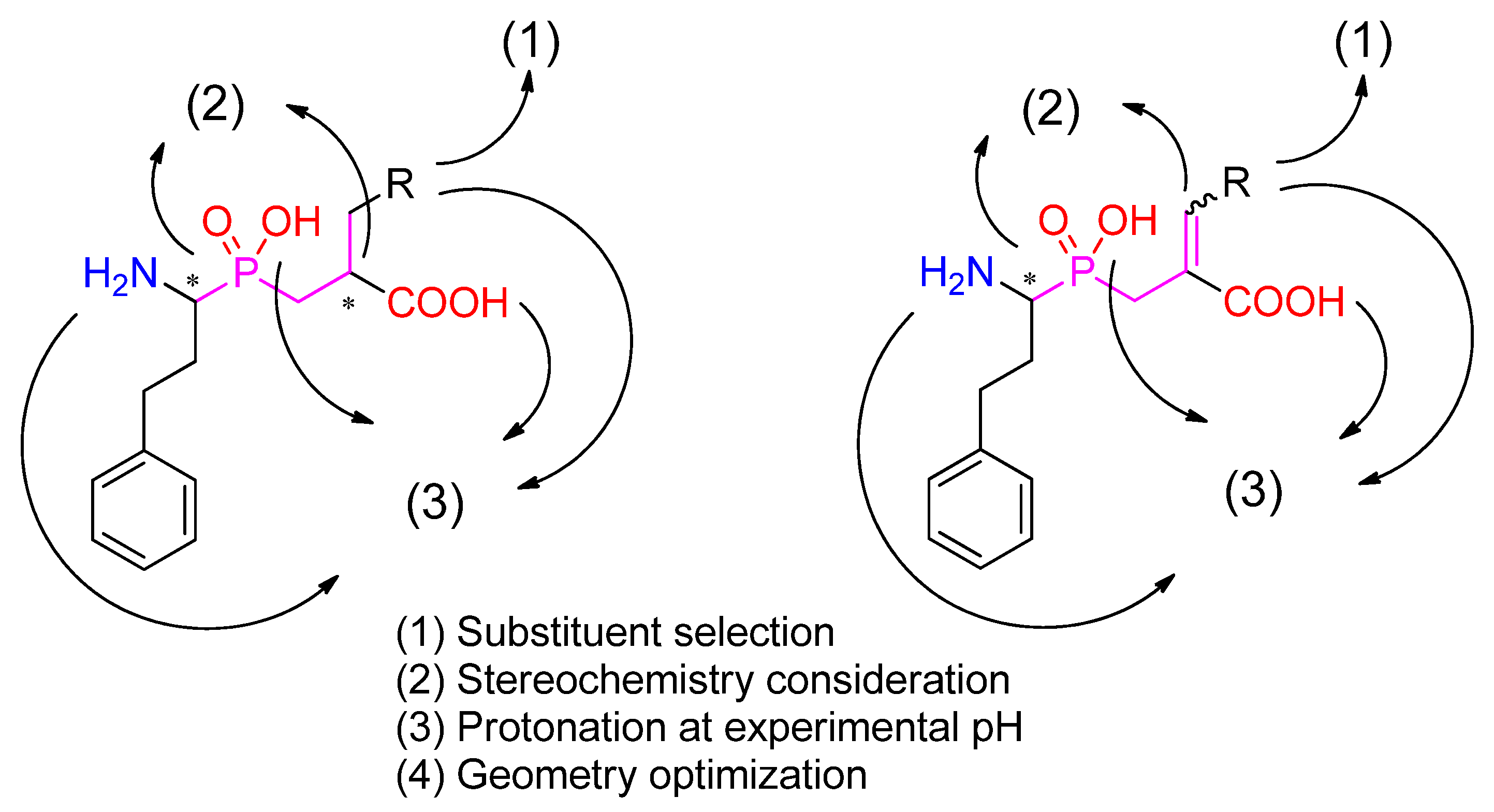
2.1. Target Selection and Preparation
2.2. Ligand Preparation
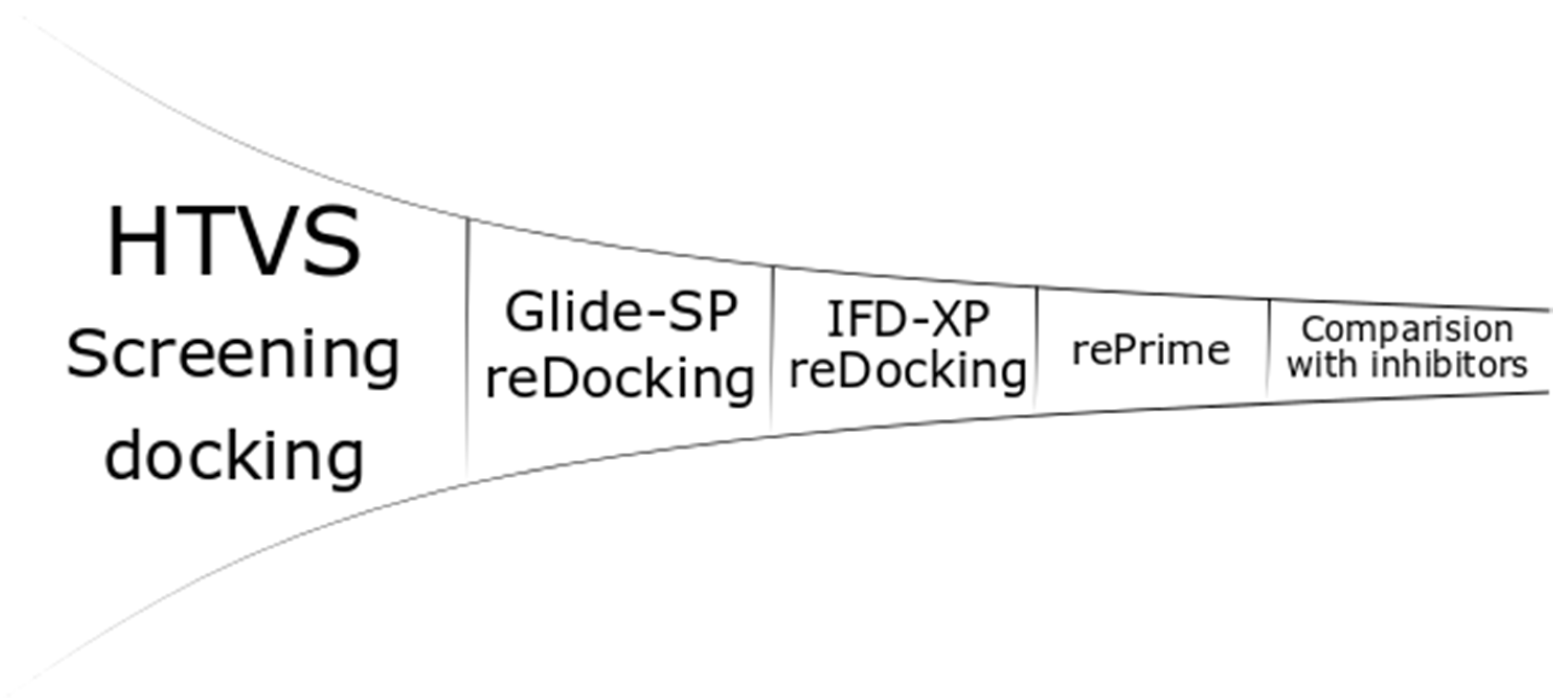
2.3. Docking Studies
2.4. ADMET Studies
3. Results and Discussion
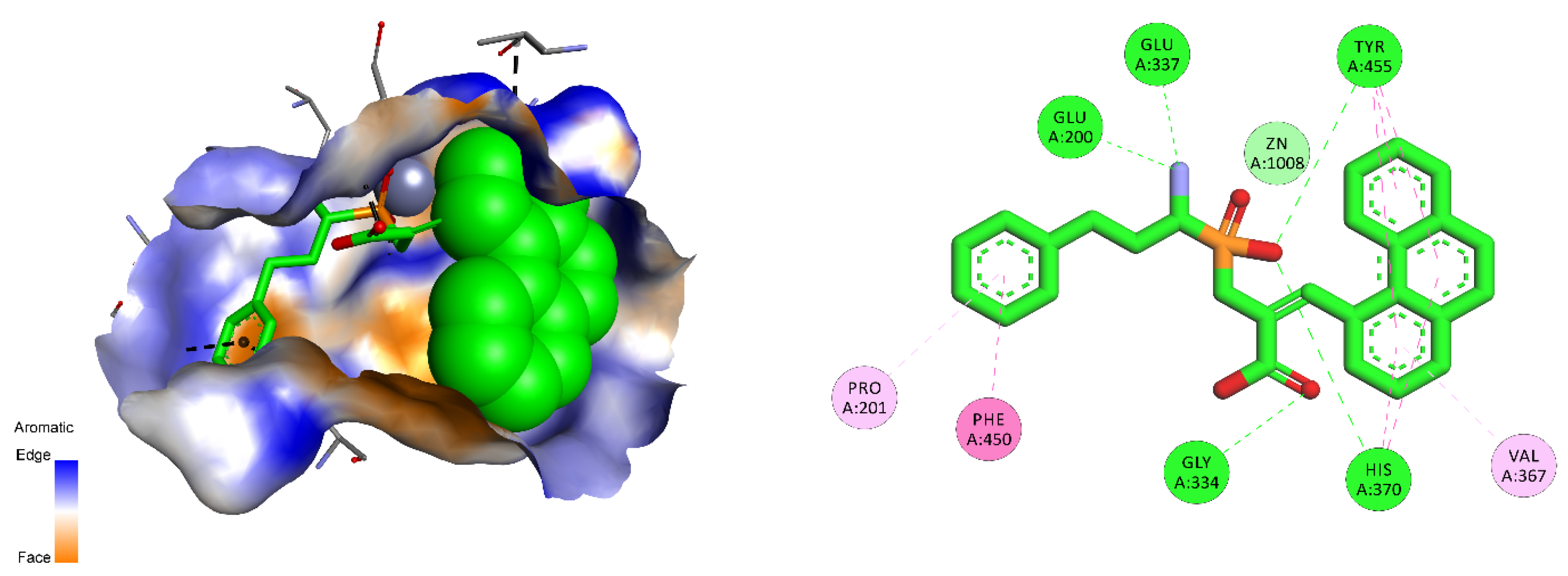
3.1. M1 Family
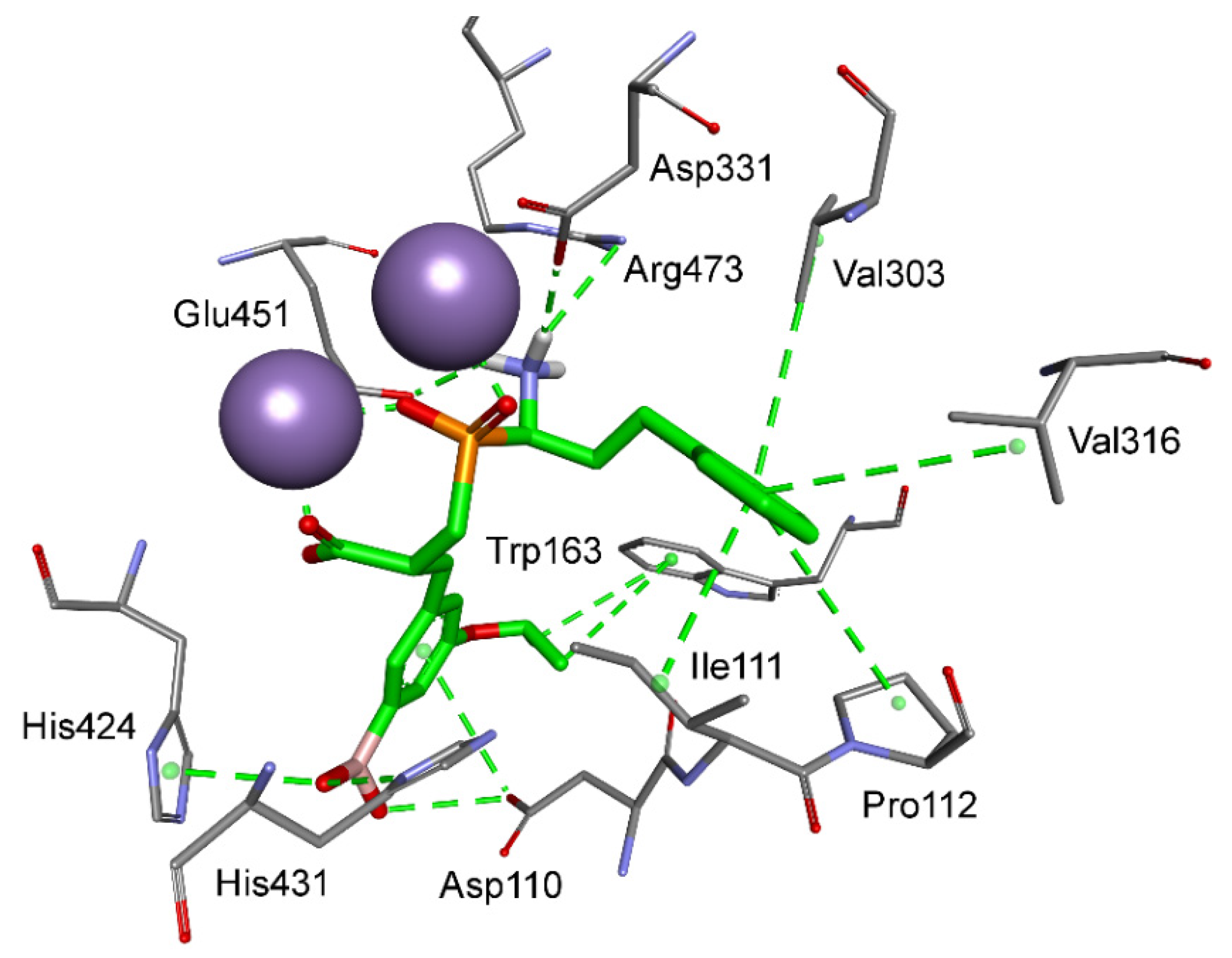
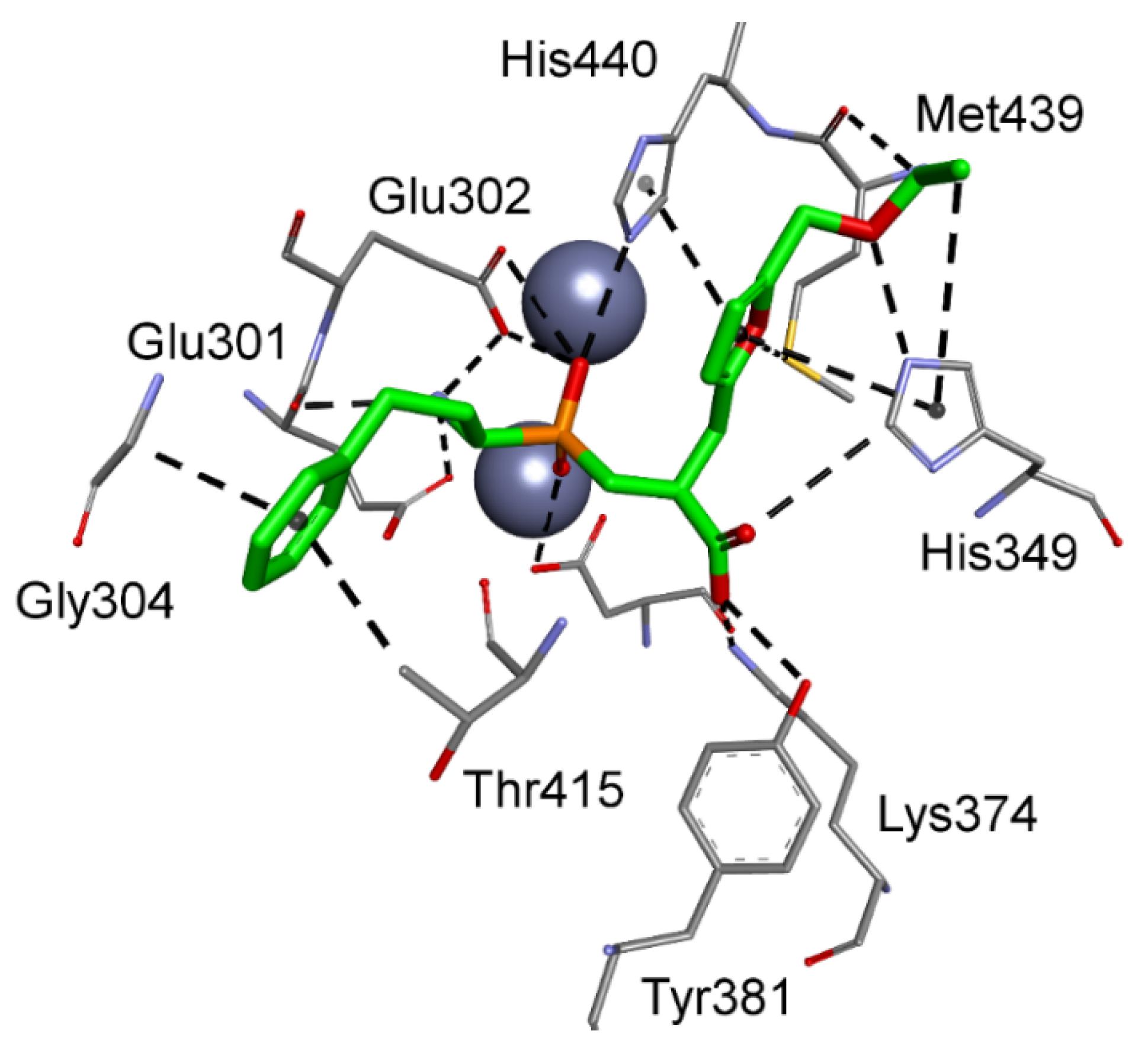
3.2. Human Aminopeptidases other than M1
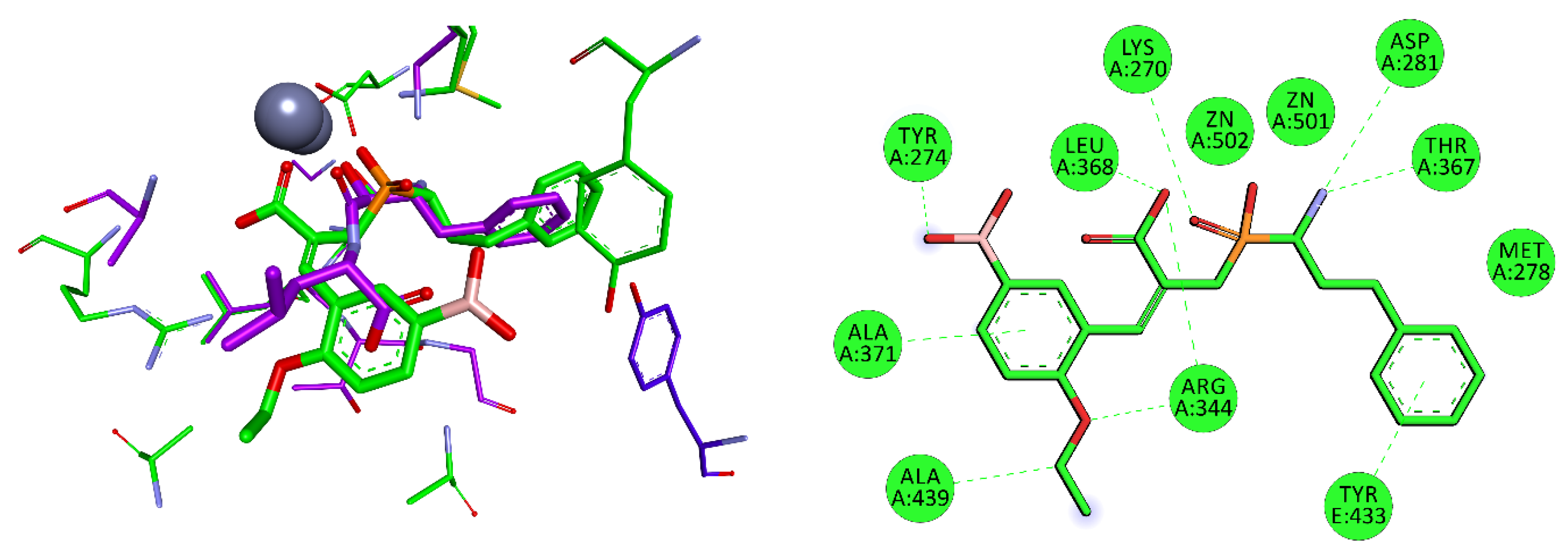
3.3. Aminopeptidases from Pathogenic Bacteria
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
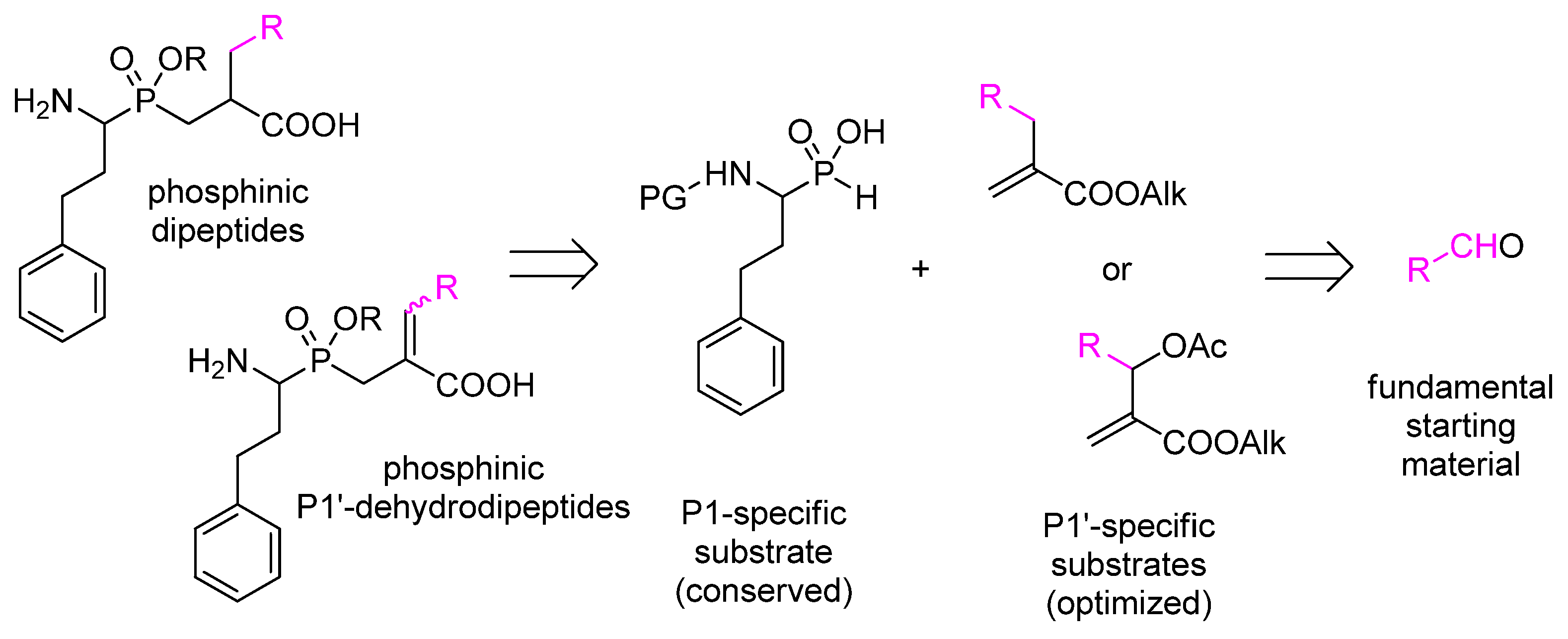
- Mucha, A. Synthesis and modifications of phosphinic dipeptide analogues. Molecules 2012, 17, 13530–13568. [Google Scholar] [CrossRef] [PubMed]
- Talma, M.; Maślanka, M.; Mucha, A. Recent developments in the synthesis and applications of phosphinic peptide analogs. Bioorg. Med. Chem. Lett. 2019, 29, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Drag, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.M.; Lendeckel, U. Aminopeptidases in Biology and Disease; Red.; Springer: Boston, MA, USA, 2004; ISBN 978-1-4613-4698-2. [Google Scholar]
- Skinner-Adams, T.S.; Stack, C.M.; Trenholme, K.R.; Brown, C.L.; Grembecka, J.; Lowther, J.; Mucha, A.; Drag, M.; Kafarski, P.; McGowan, S.; et al. Plasmodium falciparum neutral aminopeptidases: New targets for anti-malarials. Trends Biochem. Sci. 2010, 35, 53–61. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef]
- Talma, M.; Mucha, A. P-C bond formation in reactions of Morita-Baylis-Hillman adducts with phosphorus nucleophiles. Arkivoc 2016, 2017, 324–344. [Google Scholar]
- Talma, M. Phosphinic dehydrodipeptides: Diversification of the P1′ residue with the Morita–Baylis–Hillman acetates and inhibition of alanyl aminopeptidases. Int. J. Pept. Res. Ther. 2020. accepted. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.L.C. Schrödinger Release 2018-4: Schrödinger Suite 2018-4 Protein Preparation Wizard; Epik: New York, NY, USA, 2018. [Google Scholar]
- Wong, A.H.; Zhou, D.; Rini, J.M. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J. Biol. Chem. 2012, 287, 36804–36813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Lin, Y.L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, C.; Lin, Y.L.; Li, F. Structural insights into central hypertension regulation by human aminopeptidase A. J. Biol. Chem. 2013, 288, 25638–25645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stsiapanava, A.; Olsson, U.; Wan, M.; Kleinschmidt, T.; Rutishauser, D.; Zubarev, R.A.; Samuelsson, B.; Rinaldo-Matthis, A.; Haeggstrom, J.Z. Binding of Pro-Gly-Pro at the active site of leukotriene A4 hydrolase/aminopeptidase and development of an epoxide hydrolase selective inhibitor. Proc. Natl. Acad. Sci. USA 2014, 111, 4227–4232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addlagatta, A.; Gay, L.; Matthews, B.W. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry 2008, 47, 5303–5311. [Google Scholar] [CrossRef]
- Mpakali, A.; Saridakis, E.; Harlos, K.; Zhao, Y.; Kokkala, P.; Georgiadis, D.; Giastas, P.; Papakyriakou, A.; Stratikos, E. Ligand-induced conformational change of insulin-regulated aminopeptidase: Insights on catalytic mechanism and active site plasticity. J. Med. Chem. 2017, 60, 2963–2972. [Google Scholar] [CrossRef]
- Kyrieleis, O.J.P.; Goettig, P.; Kiefersauer, R.; Huber, R.; Brandstetter, H. Crystal structures of the tricorn interacting factor F3 from Thermoplasma acidophilum, a zinc aminopeptidase in three different conformations. J. Mol. Biol. 2005, 349, 787–800. [Google Scholar] [CrossRef]
- Mpakali, A.; Giastas, P.; Mathioudakis, N.; Mavridis, I.M.; Saridakis, E.; Stratikos, E. structural basis for antigenic peptide recognition and processing by endoplasmic reticulum (Er) aminopeptidase 2. J. Biol. Chem. 2015, 290, 26021–26032. [Google Scholar] [CrossRef] [Green Version]
- McGowan, S.; Porter, C.J.; Lowther, J.; Stack, C.M.; Golding, S.J.; Skinner-Adams, T.S.; Trenholme, K.R.; Teuscher, F.; Donnelly, S.M.; Grembecka, J.; et al. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc. Natl. Acad. Sci. USA 2009, 106, 2537–2542. [Google Scholar] [CrossRef] [Green Version]
- Bauvois, C.; Jacquamet, L.; Huston, A.L.; Borel, F.; Feller, G.; Ferrer, J.L. Crystal structure of the cold-active aminopeptidase from Colwellia psychrerythraea, a close structural homologue of the human bifunctional leukotriene A4 hydrolase. J. Biol. Chem. 2008, 283, 23315–23325. [Google Scholar] [CrossRef] [Green Version]
- Helgstrand, C.; Hasan, M.; Usyal, H.; Haeggstrom, J.Z.; Thunnissen, M.M.G.M. A leukotriene A(4) hydrolase-related aminopeptidase from yeast undergoes induced fit upon inhibitor binding. J. Mol. Biol. 2011, 406, 120–134. [Google Scholar] [CrossRef]
- Nocek, B.; Mulligan, R.; Bargassa, M.; Collart, F.; Joachimiak, A. Crystal structure of aminopeptidase N from human pathogen Neisseria meningitidis. Proteins 2007, 70, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strater, N.; Lipscomb, W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: Active site solvent structure and binding mode of L-leucinal, a gem-diolate transition state analogue, by X-ray crystallography. Biochemistry 1995, 34, 14792–14800. [Google Scholar] [CrossRef] [PubMed]
- Duprez, K.; Scranton, M.A.; Walling, L.L.; Fan, L. Structure of tomato wound-induced leucine aminopeptidase sheds light on substrate specificity. Acta Crystallogr. Sect. D 2014, 70, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Straeter, N.; Sherratt, D.J.; Colloms, S.D. X-Ray structure of aminopeptidase a from Escherichia coli and a model for the nucleoprotein complex in xer site-specific recombination. Embo J. 1999, 18, 4513–4522. [Google Scholar] [CrossRef] [Green Version]
- Modak, J.K.; Rut, W.; Wijeyewickrema, L.C.; Pike, R.N.; Drag, M.; Roujeinikova, A. Structural basis for substrate specificity of Helicobacter pylori M17 aminopeptidase. Biochimie 2016, 121, 60–71. [Google Scholar] [CrossRef]
- Zhan, C.; Patskovsky, Y.; Wengerter, B.C.; Ramagopal, U.; Milstein, S.; Vidal, M.; Almo, S.C. Crystal structure and function of Caenorhabditis elegans leucine aminopeptidase. To be published. Deposited in the RCSB PDB as 2HC9 in 2006.
- Kale, A.; Pijning, T.; Sonke, T.; Dijkstra, B.W.; Thunnissen, A.M. Crystal structure of the leucine aminopeptidase from Pseudomonas putida reveals the molecular basis for its enantioselectivity and broad substrate specificity. J. Mol. Biol. 2010, 398, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, S.; Huynh, K.-H.; Kang, L.W. Crystal structure of leucyl aminopeptidase (pepA) from Xoo0834, Xanthomonas oryzae pv. oryzae KACC10331. To be published. Deposited in the RCSB PDB as 3JRU in 2009.
- Chen, Y.; Farquhar, E.R.; Chance, M.R.; Palczewski, K.; Kiser, P.D. Insights into substrate specificity and metal activation of mammalian tetrahedral aspartyl aminopeptidase. J. Biol. Chem. 2012, 287, 13356–13370. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Pilka, E.S.; Riso, A.D.; Delft, F.V.; Kavanagh, K.L.; Venien-Bryan, C.; Oppermann, U.; Yue, W.W. Structure of human aspartyl aminopeptidase complexed with substrate analogue: Insight into catalytic mechanism, substrate specificity and M18 peptidase family. BMC Struct. Biol. 2012, 12, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.D.; Pandian, R.; Kim, D.; Ha, S.C.; Yoon, H.J.; Kim, K.S.; Yun, K.H.; Kim, J.H.; Kim, K.K. Structural and kinetic bases for the metal preference of the M18 aminopeptidase from Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 2014, 447, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, K.K.; Oellig, C.A.; Huynh, K.; Atkinson, S.C.; Poreba, M.; Perugini, M.A.; Trenholme, K.R.; Gardiner, D.L.; Salvesen, G.; Drag, M.; et al. X-ray crystal structure and specificity of the Plasmodium falciparum malaria aminopeptidase PfM18AAP. J. Mol. Biol. 2012, 422, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, T.; Shapiro, L. Crystal structure of aminopeptidase I from Clostridium acetobutylicum. To be published. Deposited in the RCSB PDB as 2GLJ in 2006.
- Min, T.; Shapiro, L. Crystal structure of Aminopeptidase (M18 family) from Thermotoga maritima. To be published. Deposited in the RCSB PDB as 2GLF in 2006.
- Evdokimov, A.G.; Pokross, M.; Walter, R.L.; Mekel, M.; Barnett, B.L.; Amburgey, J.; Seibel, W.L.; Soper, S.J.; Djung, J.F.; Fairweather, N.; et al. Serendipitous discovery of novel bacterial methionine aminopeptidase inhibitors. Proteins 2007, 66, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Addlagatta, A.; Quillin, M.L.; Omotoso, O.; Liu, J.O.; Matthews, B.W. Identification of an SH3-binding motif in a new class of methionine aminopeptidases from Mycobacterium tuberculosis suggests a mode of interaction with the ribosome. Biochemistry 2005, 44, 7166–7174. [Google Scholar] [CrossRef]
- Helgren, T.R.; Chen, C.; Wangtrakuldee, P.; Edwards, T.E.; Staker, B.L.; Abendroth, J.; Sankaran, B.; Housley, N.A.; Myler, P.J.; Audia, J.P.; et al. Rickettsia prowazekii methionine aminopeptidase as a promising target for the development of antibacterial agents. Bioorg. Med. Chem. 2017, 25, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Widom, J.; Kemp, C.W.; Crews, C.M.; Clardy, J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science 1998, 282, 1324–1327. [Google Scholar] [CrossRef]
- Addlagatta, A.; Matthews, B.W. Structure of the angiogenesis inhibitor ovalicin bound to its noncognate target, human type 1 methionine aminopeptidase. Protein Sci. 2006, 15, 1842–1848. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Oki, H.; Tsukihara, T.; Ogasahara, K.; Yutani, K.; Ogata, K.; Izu, Y.; Tsunasawa, S.; Kato, I. Crystal structure of methionine aminopeptidase from hyperthermophile, Pyrococcus furiosus. J. Mol. Biol. 1998, 284, 101–124. [Google Scholar] [CrossRef]
- Douangamath, A.; Dale, G.E.; D’Arcy, A.; Almstetter, M.; Eckl, R.; Frutos-Hoener, A.; Henkel, B.; Illgen, K.; Nerdinger, S.; Schulz, H.; et al. Crystal structures of Staphylococcus aureus methionine aminopeptidase complexed with keto heterocycle and aminoketone inhibitors reveal the formation of a tetrahedral intermediate. J. Med. Chem. 2004, 47, 1325–1328. [Google Scholar] [CrossRef]
- Alvarado, J.J.; Nemkal, A.; Sauder, J.M.; Russell, M.; Akiyoshi, D.E.; Shi, W.; Almo, S.C.; Weiss, L.M. Structure of a microsporidian methionine aminopeptidase type 2 complexed with fumagillin and TNP-470. Mol. Biochem. Parasitol. 2009, 168, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.C.; Bond, C.S.; Freeman, H.C.; Guss, J.M. Structural and functional implications of metal ion selection in aminopeptidase P, a metalloprotease with a dinuclear metal center. Biochemistry 2005, 44, 13820–13836. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lou, Z.; Li, X.; Zhou, W.; Ma, M.; Cao, Y.; Geng, Y.; Bartlam, M.; Zhang, X.C.; Rao, Z. Structure of human cytosolic X-prolyl aminopeptidase: A double Mn(II)-dependent dimeric enzyme with a novel three-domain subunit. J. Biol. Chem. 2008, 283, 22858–22866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Jamdar, S.N.; Goyal, V.D.; Kumar, A.; Ghosh, B.; Makde, R.D. Structure of the human aminopeptidase XPNPEP3 and comparison of its in vitro activity with Icp55 orthologs: Insights into diverse cellular processes. J. Biol. Chem. 2017, 292, 10035–10047. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, H.; Kunishima, N. Crystal structure of X-Pro aminopeptidase from Thermotoga maritima MSB8. To be published. Deposited in the RCSB PDB as 2ZSG in 2008.
- Drinkwater, N.; Sivaraman, K.K.; Bamert, R.S.; Rut, W.; Mohamed, K.; Vinh, N.B.; Scammells, P.J.; Drag, M.; McGowan, S. Structure and substrate fingerprint of aminopeptidase P from Plasmodium falciparum. Biochem. J. 2016, 473, 3189–3204. [Google Scholar] [CrossRef]
- Kumar, A.; Are, V.; Ghosh, B.; Jamdar, S.; Makde, R. R372A mutant of Xaa-Pro dipeptidase from Xanthomonas campestris at 1.85 Angstrom resolution. To be published. Deposited in the RCSB PDB as 5CDE in 2015.
- Gilboa, R.; Greenblatt, H.M.; Perach, M.; Spungin-Bialik, A.; Lessel, U.; Wohlfahrt, G.; Schomburg, D.; Blumberg, S.; Shoham, G. Interactions of Streptomyces griseus aminopeptidase with a methionine product analogue: A structural study at 1.53 A resolution. Acta Crystallogr. Sect. D 2000, 56, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Joint Center for Structural Genomics. Crystal structure of putative peptidase (NP_348812.1) from Clostridium acetobutylicum at 2.37 A resolution. To be published. Deposited in the RCSB PDB as 3K9T in 2009.
- Joint Center for Structural Genomics. Crystal structure of a hypothetical Zn-dependent exopeptidase (BDI_3547) from Parabacteroides distasonis ATCC 8503 at 1.06 A resolution. To be published. Deposited in the RCSB PDB as 3TC8 in 2011.
- Desmarais, W.; Bienvenue, D.L.; Bzymek, K.P.; Petsko, G.A.; Ringe, D.; Holz, R.C. The high-resolution structures of the neutral and the low pH crystals of aminopeptidase from Aeromonas proteolytica. J. Biol. Inorg. Chem. 2006, 11, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Akioka, M.; Nakano, H.; Tsujimoto, Y.; Matsui, H.; Nakatsu, T.; Kato, H.; Watanabe, K. 2EK8: aminopeptidase from aneurinibacillus sp. strain AM-1. To be published. Deposited in the RCSB PDB as 2EK8 in 2007.
- Odintsov, S.G.; Sabala, I.; Bourenkov, G.; Rybin, V.; Bochtler, M. Substrate access to the active sites in aminopeptidase T, a representative of a new metallopeptidase clan. J. Mol. Biol. 2005, 354, 403–412. [Google Scholar] [CrossRef]
- Odintsov, S.G.; Sabala, I.; Bourenkov, G.; Rybin, V.; Bochtler, M. Staphylococcus aureus aminopeptidases is a founding member of a new peptidase clan. J. Biol. Chem. 2005, 280, 27792–27799. [Google Scholar] [CrossRef] [Green Version]
- Badger, J.; Sauder, J.M.; Adams, J.M.; Antonysamy, S.; Bain, K.; Bergseid, M.G.; Buchanan, S.G.; Buchanan, M.D.; Batiyenko, Y.; Christopher, J.A.; et al. Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 2005, 60, 787–796. [Google Scholar] [CrossRef]
- Kim, D.; San, B.H.; Moh, S.H.; Park, H.J.; Kim, D.Y.; Lee, S.; Kim, K.K. Structural basis for the substrate specificity of PepA from Streptococcus pneumoniae, a dodecameric tetrahedral protease. Biochem. Biophys. Res. Commun. 2010, 391, 431–436. [Google Scholar] [CrossRef]
- Schoehn, G.; Vellieux, F.M.D.; Dura, M.A.; Receveur-Brechot, V.; Fabry, C.M.S.; Ruigrok, R.W.H.; Ebel, C.; Roussel, A.; Franzetti, B. An archaeal peptidase assembles into two different quaternary structures: A tetrahedron and a giant octahedron. J. Biol. Chem. 2006, 281, 36327–36337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borissenko, L.; Groll, M. Crystal structure of TET protease reveals complementary protein degradation pathways in prokaryotes. J. Mol. Biol. 2005, 346, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Dura, M.A.; Rosenbaum, E.; Larabi, A.; Gabel, F.; Vellieux, F.M.; Franzetti, B. The structural and biochemical characterizations of a novel tet peptidase complex from Pyrococcus horikoshii reveal an integrated peptide degradation system in hyperthermophilic archaea. Mol. Microbiol. 2009, 72, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Remaut, H.; Bompard-Gilles, C.; Goffin, C.; Frere, J.M.; Van Beeumen, J. Structure of the Bacillus subtilis D-aminopeptidase Dppa reveals a novel self-compartmentalizing protease. Nat. Struct. Mol. Biol. 2001, 8, 674–678. [Google Scholar] [CrossRef]
- Schrödinger LLC. Schrödinger Release 2018-4: LigPrep; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger Release 2018-4: Induced Fit; Docking Protocol; Induced Fit Docking protocol; Glide, Schrödinger, LLC: New York, NY, USA, 2016; Prime, Schrödinger, LLC: New York, NY, USA, 2018.
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinkwater, N.; Lee, J.; Yang, W.; Malcolm, T.R.; McGowan, S. M1 aminopeptidases as drug targets: Broad applications or therapeutic niche? FEBS J. 2017, 284, 1473–1488. [Google Scholar] [CrossRef]
- Peer, W.A. The role of multifunctional M1 metallopeptidases in cell cycle progression. Ann. Bot. 2011, 107, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, M.; Goto, Y.; Maruyama, M.; Hattori, A. Biochemical and enzymatic properties of the M1 family of aminopeptidases involved in the regulation of blood pressure. Heart Fail. Rev. 2008, 13, 285–291. [Google Scholar] [CrossRef]
- Mina-Osorio, P. The moonlighting enzyme CD13: Old and new functions to target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Hirose, K.; Watanabe, H. Necessity of meningococcal-glutamyl aminopeptidase for Neisseria meningitidis growth in rat cerebrospinal fluid (CSF) and CSF-like medium. J. Bacteriol. 2004, 186, 244–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Bacerio, J.; Fando, R.; Monte-Martinez, A.; Charli, J.-L.; Chávez, M. Plasmodium falciparum M1-aminopeptidase: A promising target for the development of antimalarials. Curr. Drug Targets 2014, 15, 1144–1165. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Jia, Y.; Zhang, L.; Xu, Y.; Fang, H.; Xu, W. Design, synthesis and biological evaluation of novel amino acid ureido derivatives as aminopeptidase N/CD13 inhibitors. Bioorg. Med. Chem. 2012, 20, 3807–3815. [Google Scholar] [CrossRef]
- Orning, L.; Krivi, G.; Fitzpatrick, F.A. Leukotriene A4 hydrolase. Inhibition by bestatin and intrinsic aminopeptidase activity establish its functional resemblance to metallohydrolase enzymes. J. Biol. Chem. 1991, 266, 1375–1378. [Google Scholar]
- Fournié-Zaluski, M.C.; Poras, H.; Roques, B.P.; Nakajima, Y.; Ito, K.; Yoshimoto, T. Structure of aminopeptidase N from Escherichia coli complexed with the transition-state analogue aminophosphinic inhibitor PL250. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Nikolaou, A.; Van Den Eynde, I.; Tourwé, D.; Vauquelin, G.; Tóth, G.; Mallareddy, J.R.; Poglitsch, M.; Van Ginderachter, J.A.; Vanderheyden, P.M.L. [3H]IVDE77, a novel radioligand with high affinity and selectivity for the insulin-regulated aminopeptidase. Eur. J. Pharmacol. 2013, 702, 93–102. [Google Scholar] [CrossRef]
- Papakyriakou, A.; Zervoudi, E.; Tsoukalidou, S.; Mauvais, F.-X.; Sfyroera, G.; Mastellos, D.C.; van Endert, P.; Theodorakis, E.A.; Vourloumis, D.; Stratikos, E. 3,4-Diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of M1 aminopeptidases with immune-regulating properties. J. Med. Chem. 2015, 58, 1524–1543. [Google Scholar] [CrossRef] [Green Version]
- Deprez-Poulain, R.; Flipo, M.; Piveteau, C.; Leroux, F.; Dassonneville, S.; Florent, I.; Maes, L.; Cos, P.; Deprez, B. Structure–activity relationships and blood distribution of antiplasmodial aminopeptidase-1 inhibitors. J. Med. Chem. 2012, 55, 10909–10917. [Google Scholar] [CrossRef]
- Wȩglarz-Tomczak, E.; Porȩba, M.; Byzia, A.; Berlicki, Ł.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Dra̧g, M.; Mucha, A. An integrated approach to the ligand binding specificity of Neisseria meningitidis M1 alanine aminopeptidase by fluorogenic substrate profiling, inhibitory studies and molecular modeling. Biochimie 2013, 95, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Mucha, A.; Lämmerhofer, M.; Lindner, W.; Pawełczak, M.; Kafarski, P. Individual stereoisomers of phosphinic dipeptide inhibitor of leucine aminopeptidase. Bioorg. Med. Chem. Lett. 2008, 18, 1550–1554. [Google Scholar] [CrossRef]
- Geng, N.; Zhang, W.; Li, Y.; Li, F. Aspartyl aminopeptidase suppresses proliferation, invasion, and stemness of breast cancer cells via targeting CD44. Anat. Rec. 2019, 302, 2178–2185. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.A.; Neidle, E.L.; Neidle, A. An aminopeptidase from mouse brain cytosol that cleaves N-terminal acidic amino acid residues. J. Neurochem. 1983, 40, 1727–1734. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J.; Zopf, C.J.; Huang, H.J.; Balakrishna, D.; Holub, C.; Bilakovics, J.; Fanjul, A.; Matuszkiewicz, J.; Plonowski, A.; Rolzin, P.; et al. Using target engagement biomarkers to predict clinical efficacy of MetAP2 inhibitors. J. Pharmacol. Exp. Ther. 2019, 371, 299–308. [Google Scholar] [CrossRef]
- Haldar, M.K.; Scott, M.D.; Sule, N.; Srivastava, D.K.; Mallik, S. Synthesis of barbiturate-based methionine aminopeptidase-1 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2373–2376. [Google Scholar] [CrossRef] [Green Version]
- Sulpizio, A.C.; Pullen, M.A.; Edwards, R.M.; Louttit, J.B.; West, R.; Brooks, D.P. Mechanism of vasopeptidase inhibitor-induced plasma extravasation: Comparison of omapatrilat and the novel neutral endopeptidase 24.11/angiotensin-converting enzyme inhibitor GW796406. J. Pharmacol. Exp. Ther. 2005, 315, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Prechel, M.M.; Orawski, A.T.; Maggiora, L.L.; Simmons, W.H. Effect of a new aminopeptidase P inhibitor, apstatin, on bradykinin degradation in the rat lung. J. Pharmacol. Exp. Ther. 1995, 275, 1136–1142. [Google Scholar]
- Gonzales, T.; Robert-Baudouy, J. Bacterial aminopeptidases: Properties and functions. FEMS Microbiol. Rev. 1996, 18, 319–344. [Google Scholar] [CrossRef]
- Stack, C.M.; Lowther, J.; Cunningham, E.; Donnelly, S.; Gardiner, D.L.; Trenholme, K.R.; Skinner-Adams, T.S.; Teuscher, F.; Grembecka, J.; Mucha, A.; et al. Characterization of the Plasmodium falciparum M17 leucyl aminopeptidase: A protease involved in amino acid regulation with potential for antimalarial drug development. J. Biol. Chem. 2007, 282, 2069–2080. [Google Scholar] [CrossRef] [Green Version]
- Eppinga, H.; Thio, H.B.; Schreurs, M.W.J.; Blakaj, B.; Tahitu, R.I.; Konstantinov, S.R.; Peppelenbosch, M.P.; Fuhler, G.M. Depletion of Saccharomyces cerevisiae in psoriasis patients, restored by Dimethylfumarate therapy (DMF). PLoS ONE 2017, 12, e0176955. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Armstrong, J.A.; McGechie, D.B.; Glancy, R.J. Attempt to fulfil Koch’s postulates for pyloric campylobacter. Med. J. Aust. 1985, 142, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Cheng, N.; Wang, M.W.; Zhang, J.; Shu, C.; Zhu, D.X. The leucyl aminopeptidase from Helicobacter pylori is an allosteric enzyme. Microbiology 2005, 151, 2017–2023. [Google Scholar] [CrossRef] [Green Version]
- Dutoit, R.; Brandt, N.; Legrain, C.; Bauvois, C. Functional characterization of two M42 aminopeptidases erroneously annotated as cellulases. PLoS ONE 2012, 7, e50639. [Google Scholar] [CrossRef] [Green Version]
- Leyhausen, G.; Schuster, D.K.; Vaith, P.; Zahn, R.K.; Umezawa, H.; Falke, D.; Müller, W.E.G. Identification and properties of the cell membrane bound leucine aminopeptidase interacting with the potential immunostimulant and chemotherapeutic agent bestatin. Biochem. Pharmacol. 1983, 32, 1051–1057. [Google Scholar] [CrossRef]
- Stamper, C.C.; Bienvenue, D.L.; Bennett, B.; Ringe, D.; Petsko, G.A.; Holz, R.C. Spectroscopic and X-ray crystallographic characterization of bestatin bound to the aminopeptidase from Aeromonas (Vibrio) proteolytica. Biochemistry 2004, 43, 9620–9628. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MEROPS ID | Name | Organism | PDB Code | Metal Ion(s) |
|---|---|---|---|---|
| M01.001 | Aminopeptidase N | Homo sapiens | 4FYT [14] | Zn |
| M01.001 | Aminopeptidase N | Sus scrofa | 4FKE [15] | Zn |
| M01.003 | Aminopeptidase A | Homo sapiens | 4KX7 [16] | Zn |
| M01.004 | Leukotriene A4 hydrolase | Homo sapiens | 4MKT [17] | Zn |
| M01.005 | Aminopeptidase N | Escherichia coli | 3B34 [18] | Zn |
| M01.011 | Cystinyl aminopeptidase | Homo sapiens | 5MJ6 [19] | Zn |
| M01.021 | Tricorn interacting factor F3 | Thermoplasma acidophilum | 1Z5H [20] | Zn |
| M01.024 | ERAP2 aminopeptidase | Homo sapiens | 5AB0 [21] | Zn |
| M01.029 | M1 aminopeptidase Ey | Plasmodium falciparum | 3EBH [22] | Zn |
| M01.031 | Cold-active aminopeptidase | Colwellia psychrerythraea | 3CIA [23] | Zn |
| M01.034 | Leukotriene A4 hydrolase | Saccharomyces cerevisiae | 2XQ0 [24] | Zn |
| M01.uns | Aminopeptidase N | Neisseria meningitidis | 2GTQ [25] | Zn |
| M17.001 | Leucyl aminopeptidase | Bos taurus | 1LAM [26] | Zn, Zn |
| M17.002 | Leucyl aminopeptidase | Solanum lycopersicum | 4KSI [27] | Mg, Mg |
| M17.003 | Aminopeptidase A | Escherichia coli | 1GYT [28] | Zn, Zn |
| M17.016 | Aminopeptidase A/I | Helicobacter pylori | 4ZLA [29] | Zn, Zn |
| M17.A05 | Leucyl aminopeptidase | Caenorhabditis elegans | 2HC9 [30] | Zn, Zn |
| M17.uns | Leucyl aminopeptidase | Pseudomonas putida | 3H8G [31] | Mn, Zn |
| M17.uns | Leucyl aminopeptidase | Xanthomonas oryzae | 3JRU [32] | Zn, Zn |
| M18.002 | Aspartyl aminopeptidase | Bos taurus | 3VAT [33] | Mg, Zn |
| M18.002 | Aspartyl aminopeptidase | Homo sapiens | 4DYO [34] | Zn, Zn |
| M18.002 | Aspartyl aminopeptidase | Pseudomonas aeruginosa | 4NJR [35] | Zn, Zn |
| M18.003 | Aspartyl aminopeptidase | Plasmodium falciparum | 4EME [36] | Zn, Zn |
| M18.004 | Aminopeptidase I | Clostridium acetobutylicum | 2GLJ [37] | Mn, Mn |
| M18.004 | Aminopeptidase I | Thermotoga maritima | 2GLF [38] | Mn, Mn |
| M24.001 | Methionyl aminopeptidase 1 | Escherichia coli | 2GGC [39] | Co, Co |
| M24.001 | Methionyl aminopeptidase 1 | Mycobacterium tuberculosis | 1YJ3 [40] | Co, Co |
| M24.001 | Methionyl aminopeptidase 1 | Rickettsia prowazekii | 3MX6 [41] | Mn, Mn |
| M24.002 | Methionyl aminopeptidase 2 | Homo sapiens | 1B6A [42] | Co, Co |
| M24.017 | Methionyl aminopeptidase 1 | Homo sapiens | 2GZ5 [43] | Co, Co |
| M24.035 | Methionyl aminopeptidase | Pyrococcus furiosus | 1XGS [44] | Co, Co |
| M24.036 | Methionyl aminopeptidase 1 | Staphylococcus aureus | 1QXY [45] | Co, Co, Co |
| M24A.un | Methionyl aminopeptidase 2 | Encephalitozoon cuniculi | 3FM3 [46] | Fe, Fe |
| M24.004 | Aminopeptidase P | Escherichia coli | 1WL9 [47] | Mn, Mn |
| M24.009 | Aminopeptidase P1 | Homo sapiens | 3CTZ [48] | Mn, Mn |
| M24.026 | Aminopeptidase P3 | Homo sapiens | 5 × 49 [49] | Mn, Mn |
| M24.031 | Aminopeptidase P | Thermotoga maritima | 2ZSG [50] | Zn |
| M24.038 | Aminopeptidase P | Plasmodium falciparum | 5JR6 [51] | Mn, Mn |
| M24.A11 | Aminopeptidase P | Xanthomonas campestris | 5CDE [52] | Zn, Zn |
| M28.003 | Aminopeptidase S | Streptomyces griseus | 1CP7 [53] | Zn, Zn |
| M28.uns | Non-peptidase homologues | Clostridium acetobutylicum | 3K9T [54] | Zn |
| M28.uns | Non-peptidase homologues | Parabacteroides distasonis | 3TC8 [55] | Zn |
| M28.002 | Aminopeptidase Ap1 | Vibrio proteolyticus | 1RTQ [56] | Zn, Zn |
| M28.020 | AM-1 aminopeptidase | Aneurinibacillus sp. AM-1 | 2EK8 [57] | Zn, Zn |
| M29.001 | Aminopeptidase T | Thermus thermophilus | 2AYI [58] | Zn, Zn |
| M29.005 | Aminopeptidase S | Staphylococcus aureus | 1ZJC [59] | Co, Co |
| M42.001 | Glutamyl aminopeptidase | Bacillus subtilis | 1VHE [60] | Zn |
| M42.001 | Glutamyl aminopeptidase | Streptococcus pneumoniae | 3KL9 [61] | Zn, Zn |
| M42.003 | PhTET1 aminopeptidase | Pyrococcus horikoshii | 2CF4 [62] | Co, Co |
| M42.004 | PhTET2 aminopeptidase | Pyrococcus horikoshii | 1Y0Y [63] | Zn, Zn |
| M42.009 | PhTET3 aminopeptidase | Pyrococcus horikoshii | 2VPU [64] | Zn, Zn |
| M55.001 | D-aminopeptidase DppA | Bacillus subtilis | 1HI9 [65] | Zn, Zn |
| Merops ID | Enzyme Name | P1′ | Amino Acids of S1′ |
|---|---|---|---|
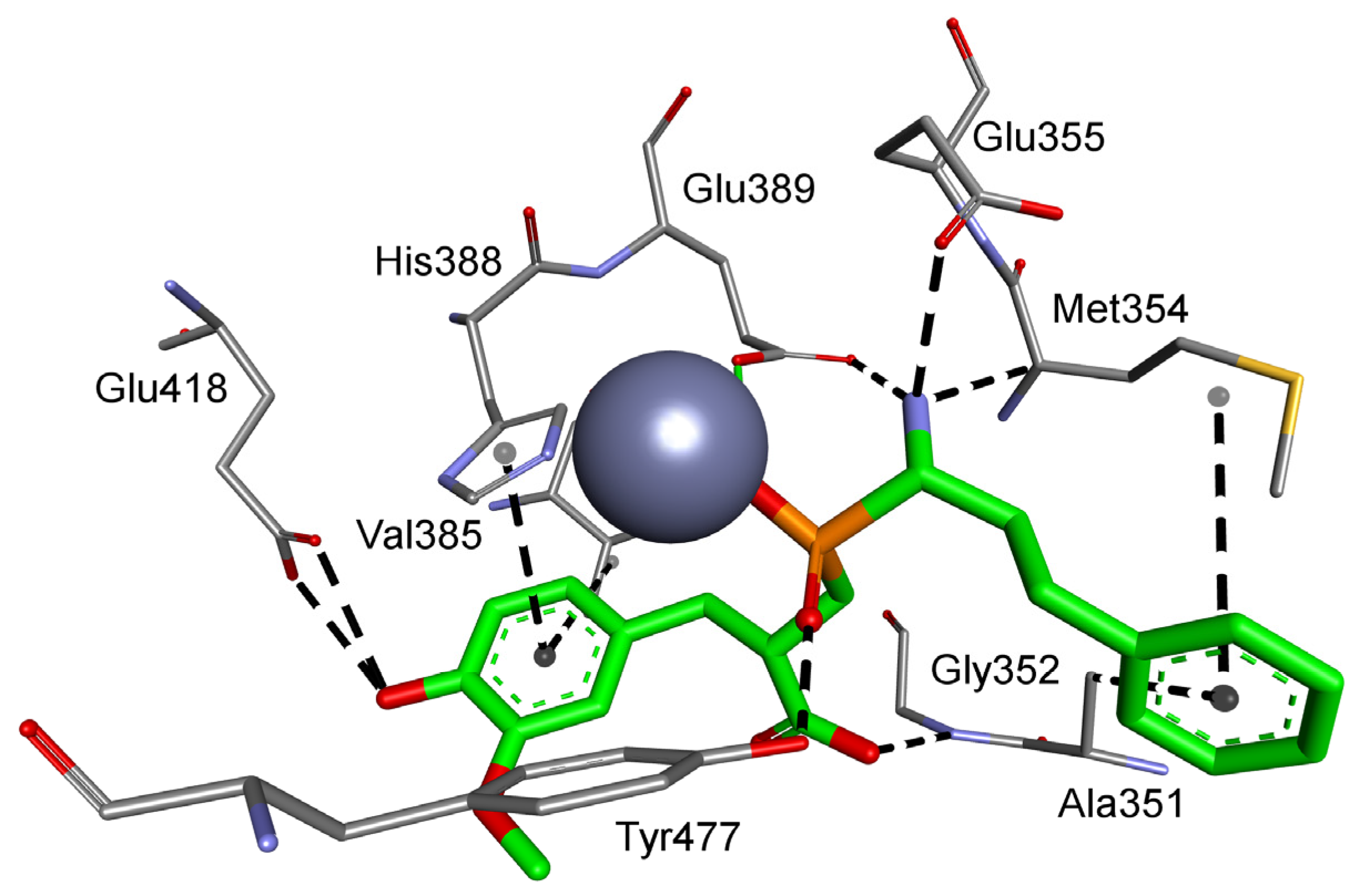
| M01.001 | Aminopeptidase N | F, G, W | Gly352, Ala353, Arg381, Thr384, Val385, His388, Glu389, Ser415, Glu418, Tyr477 |
| M01.003 | Aminopeptidase A | E, M, R | Gly357, Ala358, Asn371, Gln385, Thr389, Val390, His393, Glu394, Ser420, Glu423, Phe424, Asp444, Tyr479, Ser480 |
| M01.004 | Leukotriene A4 hydrolase | G, H | Gly268, Gly269, Asp291, Val292, His295, Glu296, Val322, Glu325, Tyr383, Arg563, Lys565 |
| M01.005 | Aminopeptidase N (bacterial) | G, N | Gly261, Ala262, Tyr275, Arg293, Val294, His297, Asp327, Tyr381, Glu382 |
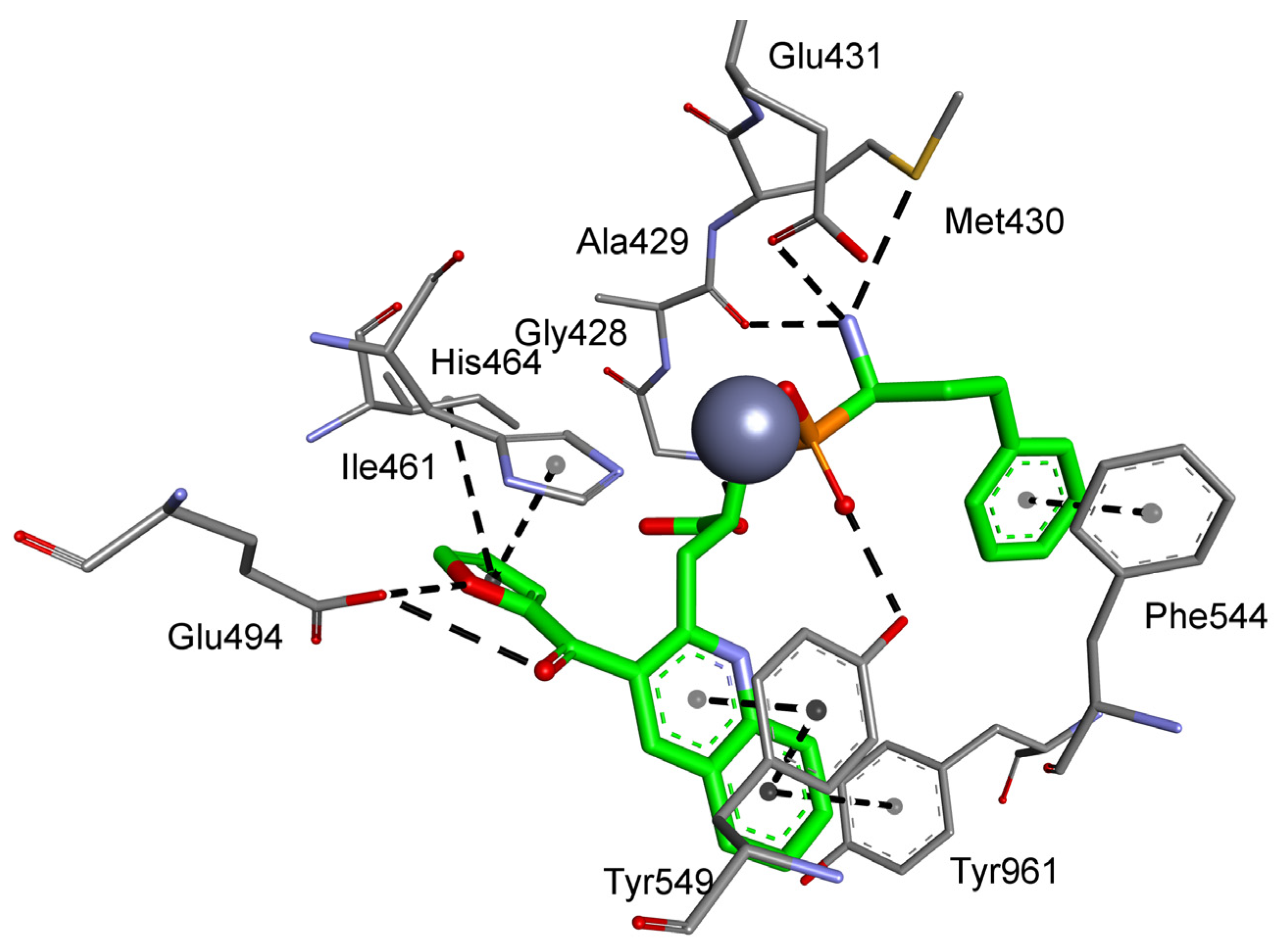
| M01.011 | Cystinyl aminopeptidase | R, T, V | Gly428, Ala429, Thr442, Leu457, Lys460, Ile461, His464, Glu465, Thr491, Glu494, Tyr549 |
| M01.024 | ERAP2 aminopeptidase | T, Y, V | Gly334, Ala335, Ser348, Trp363, Arg366, Val376, His370, Glu371, Lys397, Glu400, Tyr455 |
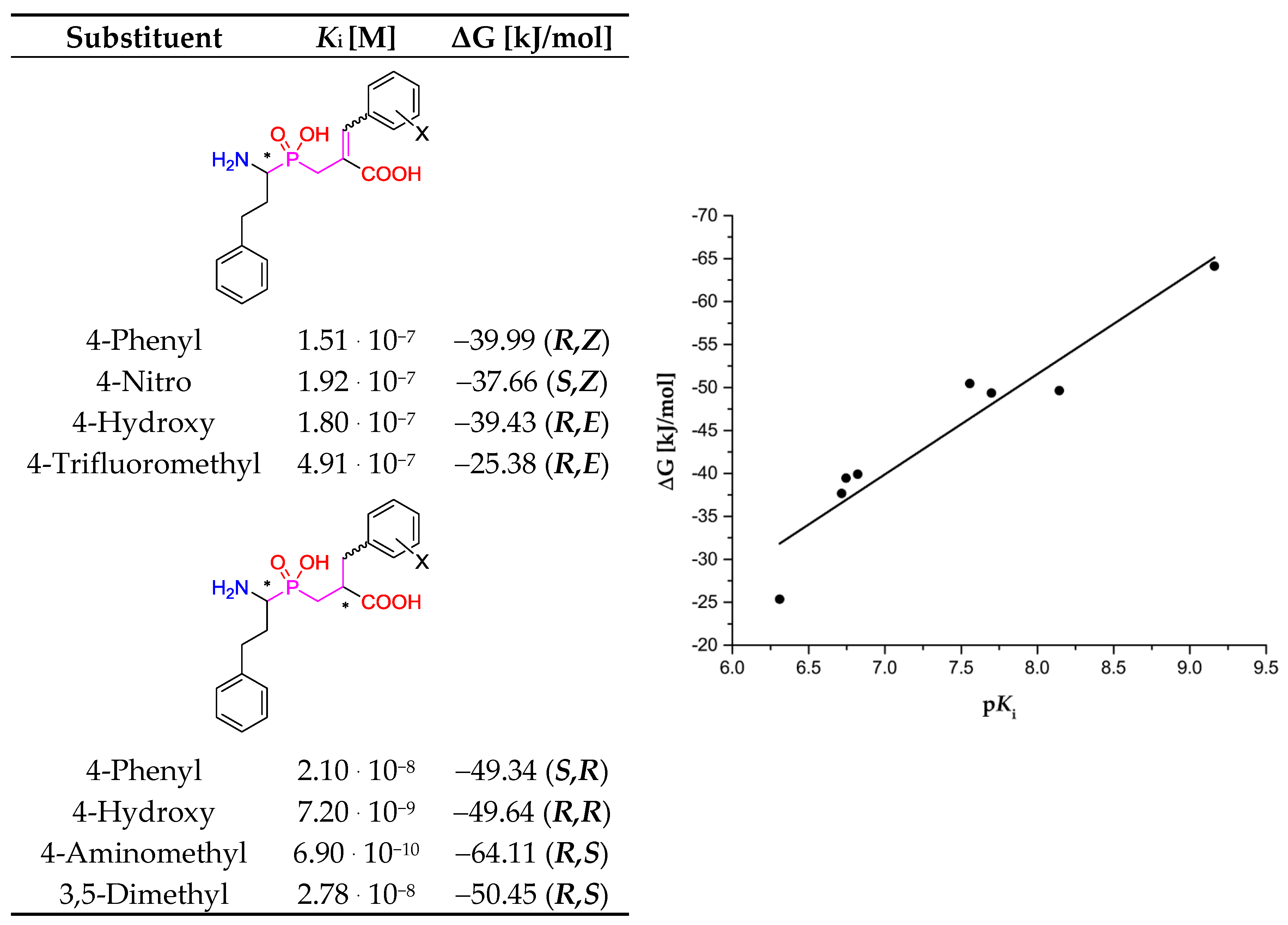
| Enzyme | Inhibitor | P1′ Substituent/Known Inhibitors | Configuration | ΔG [kJ/mol] |
|---|---|---|---|---|
| M01.001 Homo sapiens 4FYT | The best found | 4-Hydroxy-3-methoxyphenyl | S,S | −53.82 |
| Metal-interacting | ||||
| The most specific | 5-Amino-thiophen-2-yl | R,S | −34.19 | |
| Ki = 0.69 nM | R = 4-(Aminomethyl)phenyl [8] | −64.11 | ||
| M01.001 Sus scrofa 4FKE | The best found | 4-Boronophenyl MIDA ester | R,R | −39.09 |
| Metal-interacting | ||||
| The most specific | ||||
| IC50 = 8.10 µM | Bestatin [77] | −45.38 | ||
| M01.003 Homo sapiens 4KX7 | The best found | 3-Isobutylaziridin-2-yl | R,S | −38.80 |
| Metal-interacting | ||||
| The most specific | 1-Methyl-1H-imidazol-5-yl | R,S | −26.34 | |
| Ki = 75 µM | Bestatin [16] | −7.54 | ||
| M01.004 Homo sapiens 4MKT | The best found | 2-Borono-5-methoxyphenyl | R,S | −57.39 |
| Metal-interacting | none | |||
| The most specific | 2-Butyl-5-chloro-1H-imidazol-4-yl | R,S | −40.94 | |
| Ki = 200 nM | Bestatin [78] | −53.73 | ||
| M01.005 Escherichia coli 3B34 | The best found | 3,4-Dihydroxyphenyl | R,E | −49.91 |
| Metal-interacting | ||||
| The most specific | none | |||
| Ki = 1.5 nM | Ala-P[(O)(OH)CH2]-Phe-Phe [79] | −50.47 | ||
| M01.011 Homo sapiens 5MJ6 | The best found | 3-(Furan-2-carbonyl)quinolin-2-yl | R,S | −117.58 |
| Metal-interacting | ||||
| The most specific | none | |||
| Ki = 1.7 nM | IVDE771 [80] | −53.58 | ||
| M01.021 Thermoplasma acidophilum 1Z5H | The best found | 4-(6-(Hydroxymethyl)pyridin-2-yl)phenyl | R,S | −37.43 |
| Metal-interacting | 6-Ethyl-4-oxo-4H-chromen-3-yl | R,S | −37.15 | |
| The most specific | −37.15 | |||
| Unknown | Bestatin | −6.42 | ||
| M01.024 Homo sapiens 5AB0 | The best found | 2-Benzyloxyphenyl | R,S | −76.49 |
| Metal-interacting | Phenanthren-4-yl | R,Z | −36.15 | |
| The most specific | Diphenylmethyl | R,Z | −52.41 | |
| IC50 = 237 nM | DABA-Trp derivative2 [81] | −54.74 | ||
| M01.029 Plasmodium falciparum 3EBH | The best found | 3-Hydroxy-4-methoxyphenyl | R,S | −63.40 |
| Metal-interacting | ||||
| The most specific | 4-(2-Pyridyl)phenyl | R,E | −63.35 | |
| IC50 = 6 nM | BDM144713 [82] | −37.38 | ||
| M01.031 Colwellia psychrerythraea 3CIA | The best found | 2-(tert-Butylthio)phenyl | R,R | −76.28 |
| Metal-interacting | ||||
| The most specific | ||||
| Unknown value | Bestatin | −66.93 | ||
| M01.034 Saccharomyces cerevisiae 2XQ0 | The best found | 3,5-Di-tert-butylphenyl | S,R | −53.62 |
| Metal-interacting | ||||
| The most specific | 3-phenyl-1H-pyrazol-4-yl | R,R | −44.73 | |
| Unknown value | Bestatin | −47.06 | ||
| M01.un Neisseria meningitides 2GTQ | The best found | Quinolin-4-yl | R,E | −54.98 |
| Metal-interacting | ||||
| The most specific | Biphenyl-2-yl | R,E | −45.78 | |
| Ki = 54 nM | R = 4-Hydroxyphenyl [83] | −26.61 |
| Merops ID | Enzyme Name | P1′ | Amino Acids of S1′ |
|---|---|---|---|
| M18.002 | Aspartyl aminopeptidase | A, H, L | Gly123, Glu301, Glu302, Val303, Gly304, Ser305, Gly414, Thr415, His440 |
| M24.002 | Methionyl aminopeptidase 2 | A, S, V | Ala230, Asn327, Leu328, Asn329, Gly330, His331, His339, Thr343, Glu364, Phe366, Leu447 |
| M24.017 | Methionyl aminopeptidase 1 | A, S, V | Cys211, Tyr300, Gly302, His303, His310, Val315, His317, Glu336, Met338, Gln365 |
| M24.009 | Xaa-Pro aminopeptidase 1 | P | His485, His489, Glu523, Tyr527, Arg535, Glu537 |
| M24.026 | Xaa-Pro aminopeptidase 3 | A, S, P | Asp110, Trp163, Leu313, His420, His421, Val422, Gly423, His424, His431, Glu451, Pro452, Arg473 |
| Enzyme | Inhibitor | P1′ Substituent/Known Inhibitors | Configuration | ΔG [kJ/mol] |
|---|---|---|---|---|
| M18.002 Homo sapiens 4DYO | The best found | 3,4-Dibenzyloxyphenyl | R,S | −88.05 |
| The most specific | None | |||
| Metal-interacting | 5-(Ethoxymethyl)furan-2-yl | S,S | −46.49 | |
| C = 50 μg/mL – 50% | Phenantroline [87] | −16.74 | ||
| M24.002 Homo sapiens 1B6A | The best found | 4-Borono-2,3-difluorophenyl | R,S | −84.15 |
| The most specific | 3-(3-Chlorophenyl)propyl | S,S | −55.00 | |
| Metal-interacting | Phenylpropyl | S,S | −59.33 | |
| Ki = 26 nM | Beloranib [88] | −28.39 | ||
| M24.017 Homo sapiens 2GZ5 | The best found | 4-Boronophenyl | R,E | −85.18 |
| The most specific | 4-Borono-3-fluorophenyl | R,R | −51.19 | |
| Metal-interacting | 4-(6-(Hydroxymethyl)pyridinyl)phenyl | S,R | −77.96 | |
| Ki = 10 nM | Barbiturate-based inhibitor1 [89] | −53.46 | ||
| M24.009 Homo sapiens 3CTZ | The best found | 4-Borono-2-fluorophenyl | R,R | −51.91 |
| The most specific | 2-Boronophenyl | S,S | −41.61 | |
| Metal-interacting | None | |||
| Ki = 260 nM | GW7964062 [90] | −42.11 | ||
| M24.026 Homo sapiens 5 × 49 | The best found | 3-Phenylaziridin-2-yl | R,S | −102.22 |
| The most specific | 2-Borono-6-fluorophenyl | R,R | −68.93 | |
| Metal-interacting | 3-Borono-5-isopropoxyphenyl | R,R | −83.35 | |
| Ki = 640 nM | Apstatin [91] | −103.24 |
| Merops ID | Enzyme Name | P1′ | Amino Acids of S1′ |
|---|---|---|---|
| M17.016 | Aminopeptidase A/I | ND1 | Lys258, Lys270, Asn338, Asp340, Ala341, Arg344. Leu368, Thr369, Gly370, Ala371, Ile428, Gly435, Thr438, Ala439 |
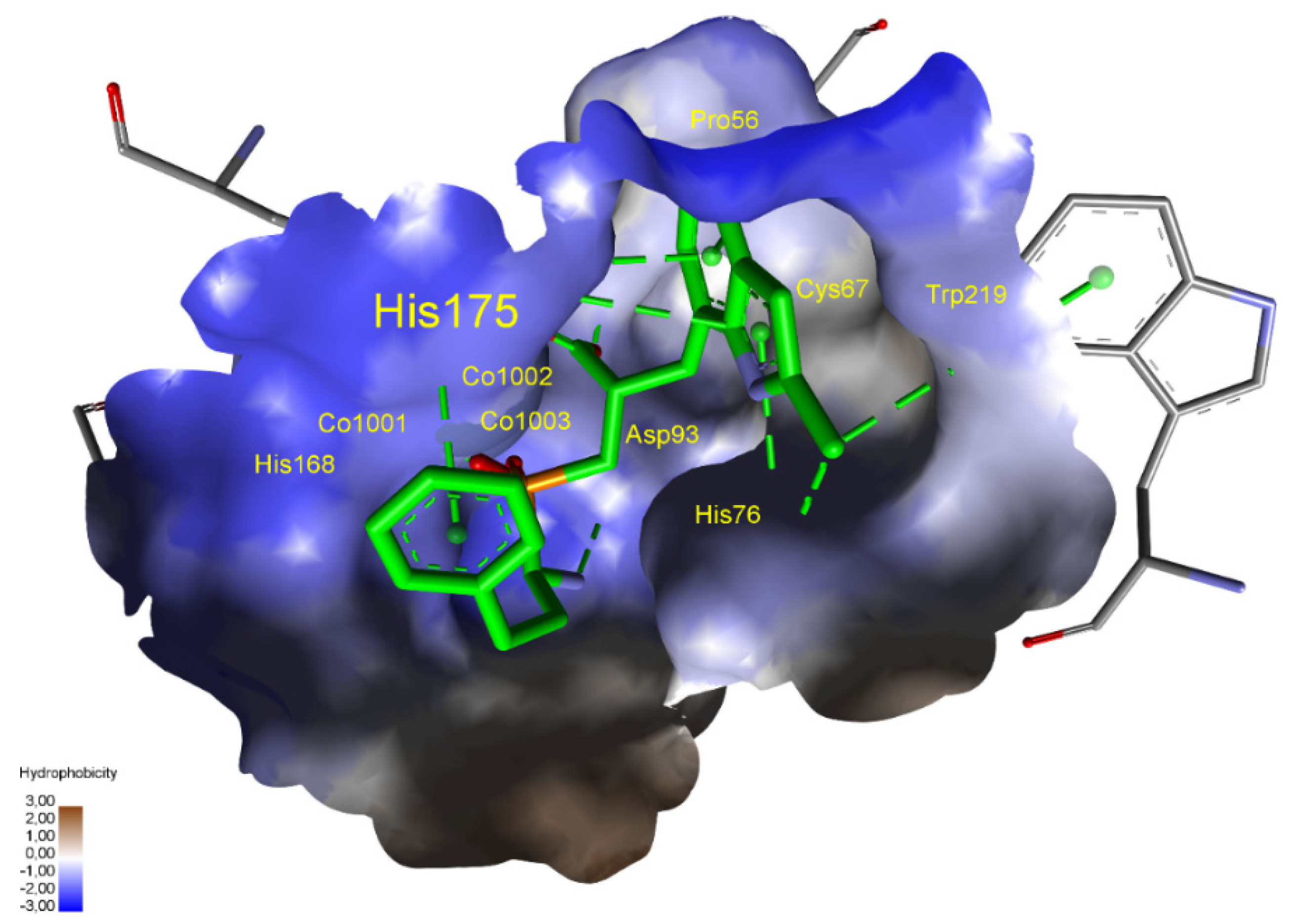
| M24.036 | Aminopeptidase S | ND1 | Ala75, Asp104, Leu165, Thr166, His168, His175, Ala179, His180, Glu202, Phe204, Phe221 |
| M42.001 | Glutamyl aminopeptidase | ND1 | Gly89, Gly90, Glu213, Glu214, Gly216, Leu217, Gly292, Thr293, His318 |
| Enzyme | Inhibitor | P1′ Substituent/Known Inhibitors | Configuration | ΔG [kJ/mol] |
|---|---|---|---|---|
| M17.016 Helicobacter pylori 4ZLA | The best found | 3-Borono-6-ethoxyphenyl | S,Z | −81.94 |
| Metal-interacting | ||||
| The most specific | None | |||
| IC50 = 49 nM | Bestatin [96] | −26.60 | ||
| M24.036 Staphylococcus aureus 1QXY | The best found | Benzo [1,3]dioxol-5-yl | R,R | −93.88 |
| Metal-interacting | ||||
| The most specific | 2-Methylindol-7-yl | R,S | −59.33 | |
| IC50 = 7 µM | 3-Amino-1-(cyclopropylamino) heptan-2-one [45] | −34.55 | ||
| M42.001 Streptococcus pneumoniae 3KL9 | The best found | 2-Chloro-5-phenylpyridin-3-yl | R,Z | −67.27 |
| Metal-interacting | ||||
| The most specific | 4-(Difluoromethoxy)-3-hydroxyphenyl | R,S | −56.17 | |
| Ki = 292 nM | Bestatin1 [97] | −41.97 |
| P1′ Group | MW | H-acc | H-don | LogP | Water Solubility | GI | Lipinski |
|---|---|---|---|---|---|---|---|
| 4-Hydroxy-3-methoxyphenyl | 406.39 | 6 | 2 | 0.5 | Moderately soluble | Low | Yes |
| 5-Amino-thiophen-2-yl | 381.41 | 4 | 2 | 0.16 | Soluble | Low | Yes |
| 4-Boronophenyl MIDA ester | 515.28 | 8 | 1 | −1.61 | Moderately soluble | Low | 1 (MW) |
| 3-Isobutylaziridin-2-yl | 368.41 | 4 | 2 | −1.13 | Soluble | High | Yes |
| 1-Methyl-1H-imidazol-5-yl | 365.36 | 4 | 2 | −0.64 | Soluble | High | Yes |
| 2-Borono-5-methoxyphenyl | 434.21 | 7 | 3 | −1.07 | Soluble | Low | Yes |
| 2-Butyl-5-chloro-1H-imidazol-4-yl | 440.88 | 5 | 2 | 1.19 | Poorly soluble | Low | Yes |
| 3,4-Dihydroxyphenyl | 390.35 | 6 | 3 | 0.12 | Soluble | Low | Yes |
| 3-(Furan-2-carbonyl)quinolin-2-yl | 505.48 | 7 | 1 | 1.62 | Poorly soluble | Low | 1 (MW) |
| 4-(6-(Hydroxymethyl)pyridin-2-yl)phenyl | 467.47 | 6 | 2 | 1.02 | Poorly soluble | Low | Yes |
| 6-Ethyl-4-oxo-4H-chromen-3-yl | 456.45 | 6 | 1 | 1.33 | Poorly soluble | Low | Yes |
| 2-Benzyloxyphenyl | 466.49 | 5 | 1 | 2.1 | Poorly soluble | High | Yes |
| Diphenylmethyl | 448.47 | 4 | 1 | 2.34 | Poorly soluble | High | Yes |
| Phenanthren-4-yl | 458.47 | 4 | 1 | 2.67 | Poorly soluble | High | Yes |
| 3-Hydroxy-4-methoxyphenyl | 406.39 | 6 | 2 | 0.38 | Moderately soluble | Low | Yes |
| 4-(2-Pyridyl)benzaldehyde | 435.43 | 5 | 1 | 1.38 | Poorly soluble | High | Yes |
| 2-(tert-Butylthio)phenyl | 448.54 | 4 | 1 | 2.09 | Poorly soluble | Low | Yes |
| 3,5-Di-tert-butylphenyl | 472.58 | 4 | 1 | 3.28 | Poorly soluble | High | Yes |
| 3-Phenyl-1H-pyrazol-4-yl | 426.43 | 5 | 2 | 0.77 | Poorly soluble | Low | Yes |
| Quinolin-4-yl | 409.39 | 5 | 1 | 1.04 | Moderately soluble | High | Yes |
| Biphenyl-2-yl | 434.44 | 4 | 1 | 2.27 | Poorly soluble | High | Yes |
| Enzyme | Ki [M] | ΔG [kJ/mol] |  |
| M01.001 | 3.03 · 10−6 [98] | −36.74 | |
| M01.001 | 4.05 · 10−6 [77] | −45.38 | |
| M01.003 | 7.50 · 10−6 [78] | −7.54 | |
| M01.004 | 2.00 · 10−7 [79] | −53.73 | |
| M01.029 | 4.81 · 10−7 [22] | −55.83 | |
| M17.001 | 9.00 · 10−9 [98] | −59.77 | |
| M28.002 | 1.80 · 10−8 [99] | −71.01 | |
| M42.001 | 2.92 · 10−7 [97] | −41.97 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talma, M.; Mucha, A. P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases. Biomolecules 2020, 10, 659. https://doi.org/10.3390/biom10040659
Talma M, Mucha A. P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases. Biomolecules. 2020; 10(4):659. https://doi.org/10.3390/biom10040659
Chicago/Turabian StyleTalma, Michał, and Artur Mucha. 2020. "P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases" Biomolecules 10, no. 4: 659. https://doi.org/10.3390/biom10040659
APA StyleTalma, M., & Mucha, A. (2020). P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases. Biomolecules, 10(4), 659. https://doi.org/10.3390/biom10040659