Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers


Abstract
:1. Introduction
2. Significance of Immunosuppressive Cells in Developing Skin Cancers
2.1. Significance of TAMs in Developing Skin Cancers
2.1.1. Chemokines from TAMs Determine Profiles of Tumor-Infiltrating Lymphocytes (TILs) in the Tumor Microenvironment
2.1.2. Angiogenic Factors from TAMs
2.2. Myeloid-Derived Suppressor Cells (MDSCs)
2.2.1. Significance of MDSCs in Developing Skin Cancers
2.2.2. MDSCs and ICIs
2.2.3. Cross-Talk between MDSCs and Other Immunosuppressive Cells
2.3. Regulatory T Cells: Tregs
2.3.1. Significance of Tregs in Developing Skin Cancers
2.3.2. Tregs and ICIs: Anti-PD1 Abs and Anti-CTLA4 Abs
2.4. TANs in Developing Skin Cancers
3. Immunosuppressive Myeloid Cells as a Target of Immunotherapy for Skin Cancer
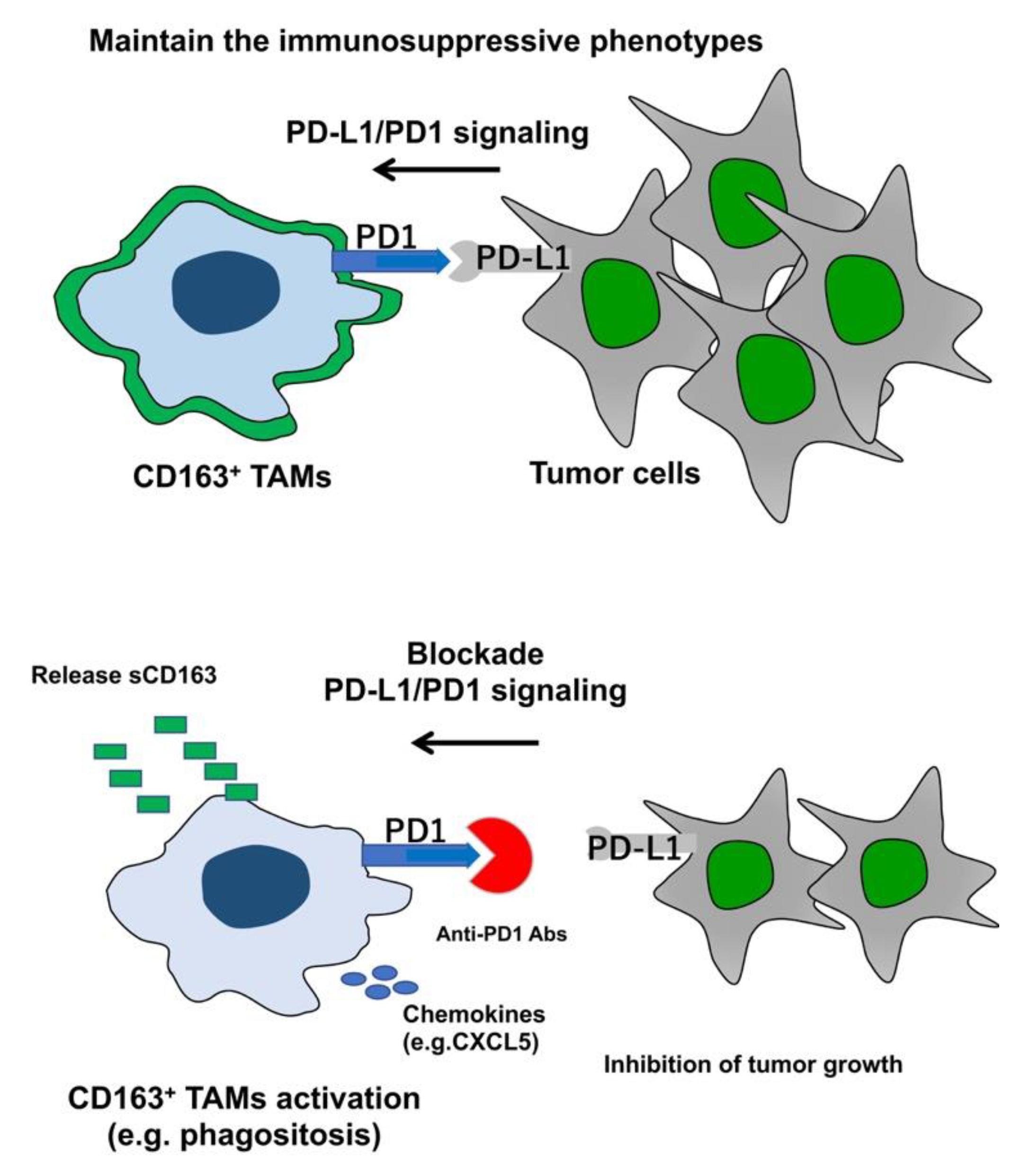
3.1. TAMs as a Target for Immunotherapy
3.2. MDSCs as a Target for Immunotherapy
4. TAM-Related Biomarkers for Predicting the Efficacy of ICIs
5. Roles of TAMs in Immune-Related Adverse Events irAEs
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fujimura, T.; Kambayashi, Y.; Fujisawa, Y.; Hidaka, T.; Aiba, S. Tumor-associated macrophages: Therapeutic targets for skin cancer. Front. Oncol. 2018, 8, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Baay, M.; Brouwer, A.; Pauwels, P.; Peeters, M.; Lardon, F. Tumor cells and tumor-associated macrophages: Secreted proteins as potential targets for therapy. Clin. Dev. Immunol. 2011, 2011, 565187–565199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, T.; Ring, S.; Umansky, V.; Mahnke, K.; Enk, A.H. Regulatory T cells (Treg) stimulate B7-H1 expression in myeloid derived suppressor cells (MDSC) in ret melanomas. J. Investig. Dermatol. 2012, 132, 1239–1246. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-associated neutrophils in cancer: Going pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.K.; Kelley, B.F.; Prokop, L.J.; Murad, M.H.; Baum, C.L. Risk factors for cutaneous squamous cell carcinoma recurrence, metastasis, and disease-specific death: A systematic review and meta-analysis. JAMA Dermatol. 2016, 152, 419–428. [Google Scholar] [CrossRef]
- Hidaka, T.; Fujimura, T.; Aiba, S. Aryl hydrocarbon receptor modulates carcinogenesis and maintenance of skin cancers. Front. Med. 2019, 6, 194. [Google Scholar] [CrossRef]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. AhR links atopic dermatitis and air pollution 1 via Artemin induction. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef]
- Wang, F.; Li, B.; Wei, Y.; Zhao, Y.; Wang, L.; Zhang, P.; Yang, J.; He, W.; Chen, H.; Jiao, Z.; et al. Tumor-derived exosomes induce PD1+ macrophage population in human gastric cancer that promotes disease progression. Oncogenesis 2018, 7, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Song, T.N.; Wang, F.R.; Yin, C.; Li, Z.; Lin, J.P.; Meng, Y.Q.; Feng, H.M.; Jing, T. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1+ TAM expansion. Oncogenesis 2019, 8, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Pan, P.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.S.; Divino, C.M.; Chen, S. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, T.; Sato, Y.; Tanita, K.; Kambayashi, Y.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; Funakoshi, T.; Hata, H.; et al. Serum soluble CD163 and CXCL5 could be predictive markers for immune related adverse event in patients with advanced melanoma treated with nivolumab. Oncotarget 2018, 9, 15542–15551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furudate, S.; Fujimura, T.; Kakizaki, A.; Kambayashi, Y.; Asano, M.; Watabe, A.; Aiba, S. The possible interaction between periostin expressed by cancer stroma and tumor-associated macrophages in developing mycosis fungoides. Exp. Dermatol. 2016, 25, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Furudate, S.; Fujimura, T.; Kakizaki, A.; Hidaka, T.; Asano, M.; Aiba, S. Tumor-associated M2 macrophages in mycosis fungoides acquire immunomodulatory function by interferon alpha and interferon gamma. J. Dermatol. Sci. 2016, 83, 182–189. [Google Scholar] [CrossRef]
- Kakizaki, A.; Fujimura, T.; Furudate, S.; Kambayashi, Y.; Yamauchi, T.; Yagita, H.; Aiba, S. Immunomodulatory effect of peritumoral administration of interferon-beta on melanoma through tumor-associated macrophages. Oncoimmunology 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Hidaka, T.; Kambayashi, Y.; Furudate, S.; Kakizaki, A.; Tono, H.; Tsukada, A.; Haga, T.; Hashimoto, A.; Morimoto, R.; et al. Phase I study of nivolumab combined with IFN-b for patients with advanced melanoma. Oncotarget 2017, 8, 71181–71187. [Google Scholar] [CrossRef] [Green Version]
- Kambayashi, Y.; Fujimura, T.; Furudate, S.; Asano, M.; Kakizaki, A.; Aiba, S. The possible interaction between receptor activator of nuclear factor kappa-B ligand (RANKL) expressed by extramammary Paget cells and its ligand on dermal macrophages. J. Investig. Dermatol. 2015, 135, 2547–2550. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J. Investig. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaiser, M.R.; Weis, C.A.; Gaiser, T.; Jiang, H.; Buder-Bakhaya, K.; Herpel, E.; Warth, A.; Xiao, Y.; Miao, L.; Brownell, I. Merkel cell carcinoma expresses the immunoregulatory ligand CD200 and induces immunosuppressive macrophages and regulatory T cells. Oncoimmunology 2018, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujimura, T.; Tanita, K.; Chunbing, L.; Matsushita, S.; Fujisawa, Y.; Otsuka, A.; Yamamoto, Y.; Hidaka, T.; Aiba, S. Malassezia-derived aryl hydrocarbon receptor ligands enhance the CCL20/Th17/soluble CD163 pathogenic axis in extra-mammary Paget’s disease. Exp. Dermatol. 2019, 28, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Werchau, S.; Toberer, F.; Enk, A.; Dammann, R.; Helmbold, P. Merkel cell carcinoma induces lymphatic microvessel formation. J. Am. Acad. Dermatol. 2012, 67, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, Y.; Zhu, P.; Zhang, D.; Liu, S.; Tang, M.; Wang, Y.; Jin, Z.; Li, D.; Yan, D.; et al. TRIM59 loss in M2 macrophages promotes melanoma migration and invasion by upregulating MMP-9 and Madcam1. Aging 2019, 11, 8623–8641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Evans, K.; Xiao, C.; DeVito, N.; Theivanthiran, B.; Holtzhausen, A.; Siska, P.J.; Blobe, G.C.; Hanks, B.A. Stromal fibroblasts mediate anti-PD-1 resistance via MMP-9 and dictate TGFβ inhibitor sequencing in melanoma. Cancer Immunol. Res. 2018, 6, 1459–1471. [Google Scholar] [CrossRef]
- Wang, T.; Ge, Y.; Xiao, M.; Lopez-Coral, A.; Azuma, R.; Somasundaram, R.; Zhang, G.; Wei, Z.; Xu, X.; Rauscher, F.J., 3rd; et al. Melanoma-derived conditioned media efficiently induce the differentiation of monocytes to macrophages that display a highly invasive gene signature. Pigment Cell Melanoma Res. 2012, 25, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Leandersson, K. The generation and identity of human myeloid-derived suppressor cells. Front. Oncol. 2020, 10, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Mengos, A.E.; Gastineau, D.A.; Gustafson, M.P. The CD14+HLA-DRlo/neg monocyte: An immunosuppressive phenotype that restrains responses to cancer immunotherapy. Front. Immunol. 2019, 10, 1147–1161. [Google Scholar] [CrossRef] [Green Version]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol. 2019, 10, 1611–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyropoulos, K.V.; Pulitzer, M.; Perez, S.; Korkolopoulou, P.; Angelopoulou, M.; Baxevanis, C.; Palomba, M.L.; Siakantaris, M. Tumor-infiltrating and circulating granulocytic myeloid-derived suppressor cells correlate with disease activity and adverse clinical outcomes in mycosis fungoides. Clin. Transl. Oncol. 2019. In Press. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Hu, S.; Wu, J.C.; Martelli, F.; Bronte, V.; Chouaib, S. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. 2015, 75, 3771–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [Green Version]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front. Immunol. 2018, 9, 2159–2168. [Google Scholar] [CrossRef]
- Han, X.; Shi, H.; Sun, Y.; Shang, C.; Luan, T.; Wang, D.; Ba, X.; Zeng, X. CXCR2 expression on granulocyte and macrophage progenitors under tumor conditions contributes to mo-MDSC generation via SAP18/ERK/STAT3. Cell Death Dis. 2019, 10, 598–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Shi, L.; Venkataramani, M.; Abdelaal, A.M.; Culpepper, C.; Kidder, K.; Liang, H.; Zen, K.; Liu, Y. Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased CXCR2 expression in mice. Eur. J. Immunol. 2018, 48, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaniego, R.; Gutiérrez-González, A.; Gutiérrez-Seijo, A.; Sánchez-Gregorio, S.; García-Giménez, J.; Mercader, E.; Márquez-Rodas, I.; Avilés, J.A.; Relloso, M.; Sánchez-Mateos, P. CCL20 expression by tumor-associated macrophages predicts progression of human primary cutaneous melanoma. Cancer Immunol. Res. 2018, 6, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Takeoka, T.; Urakawa, S.; Morimoto-Okazawa, A.; Kawashima, A.; Iwahori, K.; Takiguchi, S.; Nishikawa, H.; Sato, E.; Sakaguchi, S.; et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int. J. Cancer 2017, 140, 686–695. [Google Scholar] [CrossRef]
- Walker, L.S.; Sansom, D.M. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015, 36, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Gambichler, T.; Schröter, U.; Höxtermann, S.; Susok, L.; Stockfleth, E.; Becker, J.C. Decline of programmed death-1-positive circulating T regulatory cells predicts more favourable clinical outcome of patients with melanoma under immune checkpoint blockade. Br. J. Dermatol. 2020, 182, 1214–1220. [Google Scholar] [CrossRef]
- Gambichler, T.; Schröter, U.; Höxtermann, S.; Susok, L.; Stockfleth, E.; Becker, J.C. A brief communication on circulating PD-1-positive T-regulatory lymphocytes in melanoma patients undergoing adjuvant immunotherapy with nivolumab. J. Immunother. 2019, 42, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Schoonderwoerd, M.J.; Koops, M.F.; Angela, R.A.; Koolmoes, B.; Toitou, M.; Paauwe, M.; Barnhoorn, M.C.; Liu, Y.; Sier, C.F.; Hardwick, J.C.; et al. Targeting endoglin expressing regulatory T cells in the tumor microenvironment enhances the effect of PD1 checkpoint inhibitor immunotherapy. Clin. Cancer Res. 2020. In Press. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsthuber, A.; Lipp, K.; Andersen, L.; Ebersberger, S.; Graña-Castro, O.; Ellmeier, W.; Petzelbauer, P.; Lichtenberger, B.M.; Loewe, R. CXCL5 as regulator of neutrophil function in cutaneous melanoma. J. Investig. Dermatol. 2019, 139, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonska, J.; Wu, C.F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Simon, N.; Antignani, A.; Hewitt, S.M.; Gadina, M.; Alewine, C.; FitzGerald, D. Tofacitinib enhances delivery of antibody-based therapeutics to tumor cells through modulation of inflammatory cells. JCI Insight 2019, 4, 1–18. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Neutrophils in cancer: Two sides of the same coin. J. Immunol. Res. 2015, 2015, 983698–983719. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Wu, L.; Chen, X.; Zhao, J.; Martin, B.; Zepp, J.A.; Ko, J.S.; Gu, C.; Cai, G.; Ouyang, W.; Sen, G.; et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J. Exp. Med. 2015, 212, 1571–1587. [Google Scholar] [CrossRef] [PubMed]
- Gasparoto, T.H.; de Oliveira, C.E.; de Freitas, L.T.; Pinheiro, C.R.; Ramos, R.N.; da Silva, A.L.; Cavassani, K.A.; Campanelli, A.P. Inflammatory events during murine squamous cell carcinoma development. J. Inflamm. 2012, 9, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Pascual-García, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martínez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8+ T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416–2427. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, T.; Okuyama, R.; Ohtani, T.; Ito, Y.; Haga, T.; Hashimoto, A.; Aiba, S. Perilesional treatment of metastatic melanoma with interferon-beta. Clin. Exp. Dermatol. 2009, 34, 793–799. [Google Scholar] [CrossRef]
- Antsiferova, M.; Piwko-Czuchra, A.; Cangkrama, M.; Wietecha, M.; Sahin, D.; Birkner, K.; Amann, V.C.; Levesque, M.; Hohl, D.; Dummer, R.; et al. Activin promotes skin carcinogenesis by attraction and reprogramming of macrophages. EMBO Mol. Med. 2017, 9, 27–45. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974–3990. [Google Scholar] [CrossRef]
- Nam, S.; Kang, K.; Cha, J.S.; Kim, J.W.; Lee, H.G.; Kim, Y.; Yang, Y.; Lee, M.S.; Lim, J.S. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J. Leukoc. Biol. 2016, 100, 1273–1284. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity 2017, 47, 284–297. [Google Scholar] [CrossRef]
- Sierra, R.A.; Trillo-Tinoco, J.; Mohamed, E.; Yu, L.; Achyut, B.R.; Arbab, A.; Bradford, J.W.; Osborne, B.A.; Miele, L.; Rodriguez, P.C. Anti-Jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res. 2017, 77, 5628–5638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol. Immunother. 2017, 66, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Han, X.; Sun, Y.; Shang, C.; Wei, M.; Ba, X.; Zeng, X. Chemokine (C-X-C motif) ligand 1 and CXCL2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci. 2018, 109, 3826–3839. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, Y.; Fujimura, T.; Hidaka, T.; Aiba, S. Biomarkers for the prediction of efficacies of anti-PD1 antibodies: Mini reviews. Front. Med. 2019, 6, 174. [Google Scholar] [CrossRef] [Green Version]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Ho, W.J.; Yarchoan, M.; Hopkins, A.; Mehra, R.; Grossman, S.; Kang, H. Association between pretreatment lymphocyte count and response to PD1 inhibitors in head and neck squamous cell carcinomas. J. Immunother. Cancer 2018, 6, 84–92. [Google Scholar] [CrossRef]
- Fujisawa, Y.; Yoshino, K.; Otsuka, A.; Funakoshi, T.; Fujimura, T.; Yamamoto, Y.; Hata, H.; Tanaka, R.; Yamaguchi, K.; Nonomura, Y.; et al. Baseline neutrophil to lymphocyte ratio and serum LDH level associated with outcome of nivolumab immunotherapy in Japanese advanced melanoma population. Br. J. Dermatol. 2018, 179, 213–215. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Yagi, T.; Baba, Y.; Okadome, K.; Kiyozumi, Y.; Hiyoshi, Y.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Watanabe, M.; et al. Tumour-associated macrophages are associated with poor prognosis and programmed death ligand 1 expression in oesophageal cancer. Eur. J. Cancer 2019, 111, 38–49. [Google Scholar] [CrossRef]
- Fujimura, T.; Sato, Y.; Tanita, K.; Kambayash, Y.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; Funakoshi, T.; Hata, H.; et al. Serum level of soluble CD163 may be a predictive marker of the effectiveness of nivolumab in patients with advanced cutaneous melanoma. Front. Oncol. 2018, 8, 530–538. [Google Scholar] [CrossRef]
- Fujimura, T.; Sato, Y.; Tanita, K.; Lyu, C.; Kambayash, Y.; Amagai, R.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; et al. Association of baseline serum levels of CXCL5 with the efficacy of nivolumab in advanced melanoma. Front. Med. 2019, 6, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, T.; Tanita, K.; Sato, Y.; Lyu, C.; Kambayashi, Y.; Fujisawa, Y.; Uchi, H.; Yamamoto, Y.; Otsuka, A.; Yoshino, K.; et al. Immune checkpoint inhibitor-induced vitiligo in advanced melanoma could be related to increased levels of CCL19. Br. J. Dermatol. 2020, 182, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Darnell, E.P.; Mooradian, M.J.; Baruch, E.N.; Yilmaz, M.; Reynolds, K.L. Immune-related adverse events (irAEs): Diagnosis, management, and clinical pearls. Curr. Oncol. Rep. 2020, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Furudate, S.; Kakizaki, A.; Hidaka, T.; Haga, T.; Hashimoto, A.; Morimoto, R.; Aiba, S. Isolated ACTH deficiency possibly caused by nivolumab in a metastatic melanoma patient. J. Dermatol. 2017, 44, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Kambayashi, Y.; Tanita, K.; Sato, Y.; Hidaka, T.; Otsuka, A.; Tanaka, H.; Furudate, S.; Hashimoto, A.; Aiba, S. Two cases of Vogt-Koyanagi Harada disease-like uveitis developing from an advanced melanoma patient treated by sequential administration of nivolumab and dabrafenib/trametinib therapy. J. Dermatol. 2018, 45, 735–737. [Google Scholar] [CrossRef]
- Nelson, C.A.; Singer, S.; Chen, T.; Puleo, A.E.; Lian, C.G.; Wei, E.X.; Giobbie-Hurder, A.; Mostaghimi, A.; LeBoeuf, N.R. Bullous pemphigoid after anti-PD-1 therapy: A retrospective case-control study evaluating impact on tumor response and survival outcomes. J. Am. Acad. Dermatol. 2020. In Press. [Google Scholar] [CrossRef]
- Voudouri, D.; Nikolaou, V.; Laschos, K.; Charpidou, A.; Soupos, N.; Triantafyllopoulou, I.; Panoutsopoulou, I.; Aravantinos, G.; Syrigos, K.; Stratigos, A. Anti-PD1/PDL1 induced psoriasis. Curr. Probl. Cancer 2017, 41, 407–412. [Google Scholar] [CrossRef]
- Yilmaz, M.; Mese, S.G.; Celik, U. Nivolumab-induced lichen planus. J. Oncol. Pharm. Pract. 2019, 23, 758–760. [Google Scholar] [CrossRef]
- Tojo, G.; Fujimura, T.; Kawano, M.; Ogasawara, K.; Kambayashi, Y.; Furudate, S.; Mizuashi, M.; Aiba, S. Comparison of IL-17 producing cells in different clinical types of alopecia areata. Dermatology 2013, 227, 77–82. [Google Scholar] [CrossRef]
- Furudate, S.; Fujimura, T.; Kambayashi, Y.; Kakizaki, A.; Aiba, S. Comparison of CD163+ CD206+ M2 macrophages in the lesional skin of bullous pemphigoid and pemphigus vulgaris: The possible pathogenesis of bullous pemphigoid. Dermatology 2014, 229, 369–378. [Google Scholar] [CrossRef]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Aiba, S. A possible interaction between periostin and CD163+ skin-resident macrophages in pemphigus vulgaris and bullous pemphigoid. Exp. Dermatol. 2017, 26, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Riani, M.; Muller, C.; Bour, C.; Bernard, P.; Antonicelli, F.; Le Jan, S. Blister fluid induces MMP-9-associated M2-type macrophages in bullous pemphigoid. Front. Immunol. 2019, 10, 1858–1869. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Duculan, J.; Suárez-Fariñas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef] [Green Version]
- Van Gorp, H.; Delputte, P.L.; Nauwynck, H.J. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol. Immunol. 2010, 47, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Keller, M.; Kim, Y.; Cronin, H.; Richards, A.; Gibney, G.; Weber, J.S. Nivolumab in resected and unresectable metastatic melanoma: Characteristics of immune-related adverse events and association with outcomes. Clin. Cancer Res. 2016, 22, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, T.; Kambayashi, Y.; Sato, T.; Tanita, K.; Amagai, R.; Hashimoto, A.; Hidaka, T.; Aiba, S. Successful treatment of unresectable advanced melanoma with pre-surgical administration of nivolumab with ipilimumab. Front. Med. 2019, 6, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, M.; Sugihara, S.; Mori, S.; Hamada, K.; Sasaki, Y.; Yoshikawa, S.; Kiyohara, Y. Immune-related adverse events correlate with improved survival in patients with advanced mucosal melanoma treated with nivolumab: A single-center retrospective study in Japan. J. Dermatol. 2020. In Press. [Google Scholar] [CrossRef]
- Okada, N.; Kawazoe, H.; Takechi, K.; Matsudate, Y.; Utsunomiya, R.; Zamami, Y.; Goda, M.; Imanishi, M.; Chuma, M.; Hidaka, N.; et al. Association between immune-related adverse events and clinical efficacy in patients with melanoma treated with nivolumab: A multicenter retrospective study. Clin. Ther. 2019, 41, 59–67. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Subtypes | Positive | Negative | |
|---|---|---|---|
| TAMs | M1 | CD68, CD86, CD169, HLA-DR, CCR7 | |
| M2 | CD163, CD204, CD206, PD-L1, ARG1 | ||
| MDSCs | MoMDSC | CD11b, PGE2, IL-10, TGFb, iNOS, ARG1 | HLA-DR, CD14 |
| G-MDSC | CD15, CD33, CD66b, ROS, G-CSF, ARG1 | HLA-DR, CD14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimura, T.; Aiba, S. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules 2020, 10, 1087. https://doi.org/10.3390/biom10081087
Fujimura T, Aiba S. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules. 2020; 10(8):1087. https://doi.org/10.3390/biom10081087
Chicago/Turabian StyleFujimura, Taku, and Setsuya Aiba. 2020. "Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers" Biomolecules 10, no. 8: 1087. https://doi.org/10.3390/biom10081087
APA StyleFujimura, T., & Aiba, S. (2020). Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules, 10(8), 1087. https://doi.org/10.3390/biom10081087