8-Deoxy-Rifamycin Derivatives from Amycolatopsis mediterranei S699 ΔrifT Strain

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. DNA Manipulation
2.2.1. Gene Knock-Out
2.2.2. Gene Complementation
2.3. Detection and Analysis of the Metabolites in Mutants
2.4. General Experimental Procedures
2.5. Extraction and Isolation
2.6. Antimicrobial Assay
2.7. Cytotoxicity Assay
2.8. Anti-Type III Secretion System (T3SS) Assay
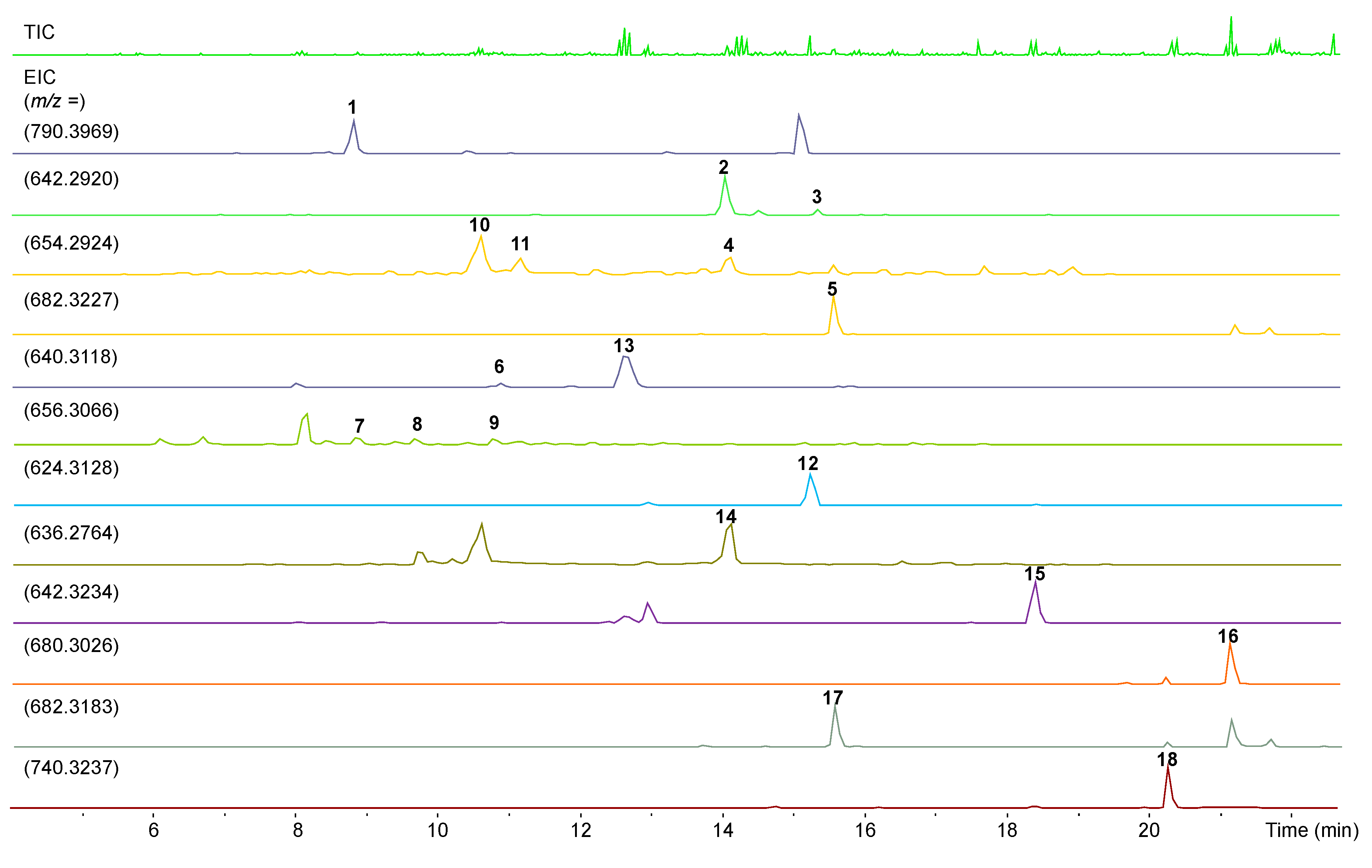
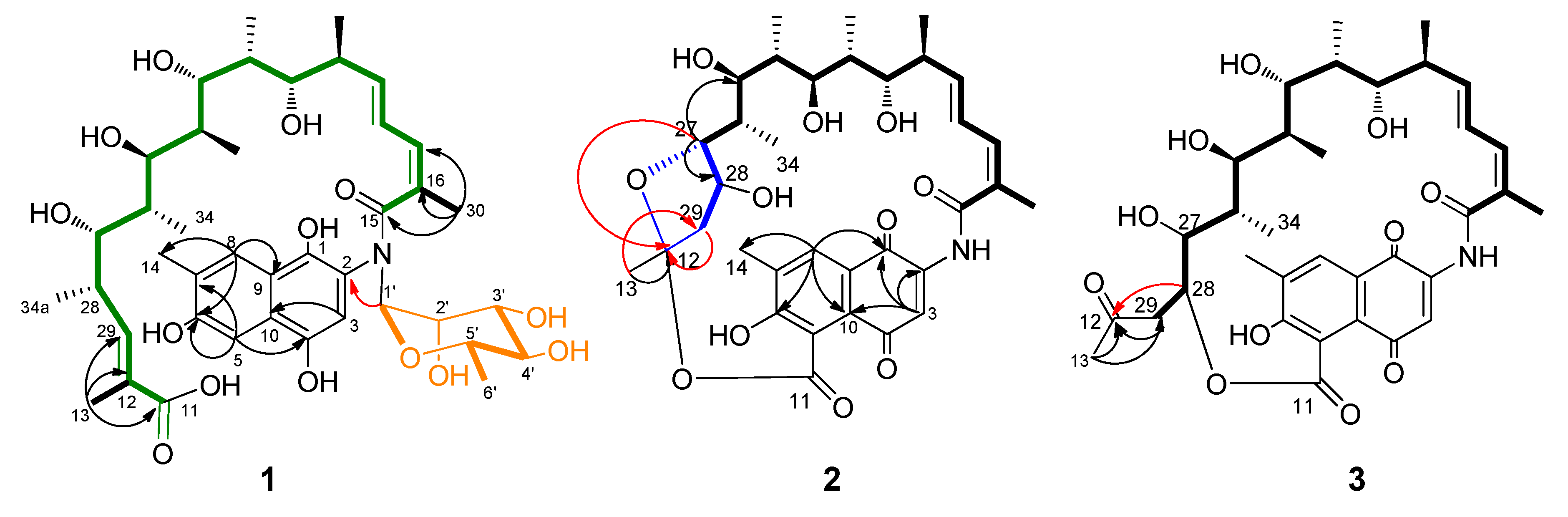
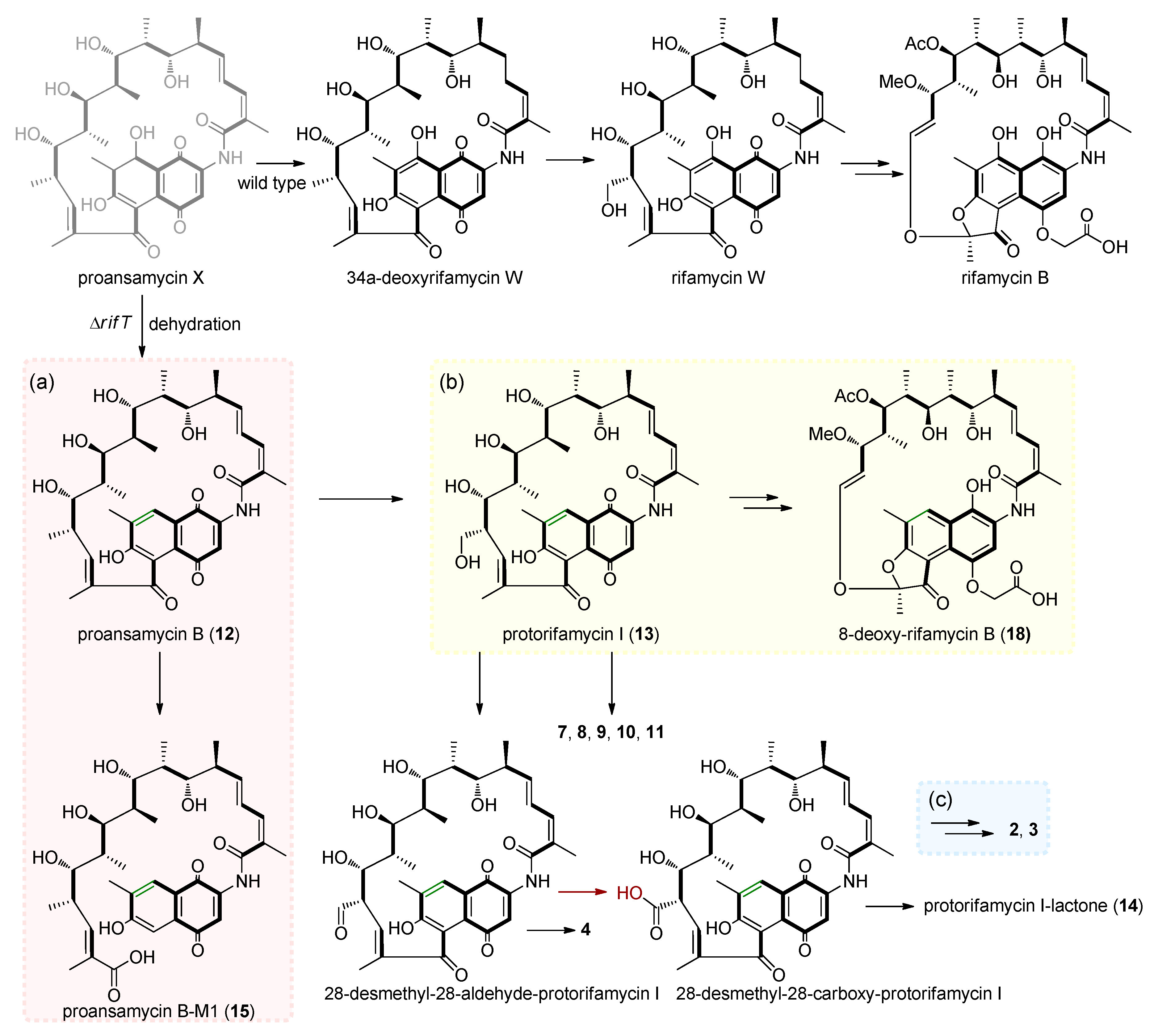
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rinehart, K.L.; Shield, L.S. Chemistry of the ansamycin antibiotics. In Fortschritte der Chemie Organischer Naturstoffe; Springer: Vienna, Austria, 1976; Volume 33, pp. 231–307. [Google Scholar]
- Wehrli, W. Ansamycins chemistry, biosynthesis and biological activity. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 1977; Volume 72, pp. 21–49. [Google Scholar]
- Maggi, N.; Pasqualucci, C.R.; Ballotta, R.; Sensi, P. Rifampicin: A new orally active rifamycin. Chemotherapy 1966, 11, 285–292. [Google Scholar] [CrossRef]
- Cassady, J.M.; Chan, K.K.; Floss, H.G.; Leistner, E. Recent developments in the maytansinoid antitumor agents. Chem. Pharm. Bull. 2004, 52, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Kusari, P.; Kusari, S.; Eckelmann, D.; Zühlke, S.; Kayser, O.; Spiteller, M. Cross-species biosynthesis of maytansine in Maytenus serrata. RSC Adv. 2016, 6, 10011–10016. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [Green Version]
- Kang, Q.; Shen, Y.; Bai, L. Biosynthesis of 3,5-AHBA-derived natural products. Nat. Prod. Rep. 2012, 29, 243–263. [Google Scholar] [CrossRef]
- Sensi, P.; Margalith, P.; Timbal, M.T. Rifamycin, a new antibiotic. Farmaco. Sci. 1959, 14, 146–147. [Google Scholar]
- Sensi, P.; Greco, A.M.; Gallo, G.G.; Rolland, G. Isolation and structure determination of a new amicetin-like antibiotic: Amicetin B. Antibiot. Chemother. 1957, 7, 645–652. [Google Scholar]
- Sensi, P. Applications of paper chromatography & countercurrent distribution to steroids & antibiotics. Boll. Chim. Farm. 1957, 96, 437–457. [Google Scholar]
- Wehrli, W.; Staehelin, M. Actions of the rifamycins. Bacteriol. Rev. 1971, 35, 290–309. [Google Scholar] [CrossRef]
- Steffen, R.; Jiang, Z.D.; Gracias Garcia, M.L.; Araujo, P.; Stiess, M.; Nacak, T.; Greinwald, R.; DuPont, H.L. Rifamycin SV-MMX(R) for treatment of travellers’ diarrhea: Equally effective as ciprofloxacin and not associated with the acquisition of multi-drug resistant bacteria. J. Travel Med. 2018, 25, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cho, N.K.; Sunada, Y.; Nohara, S. Clinical studies on a new antibiotic, rifamycin SV. J. Showa Med Assoc. 1963, 23, 86–87. [Google Scholar]
- Girling, D.J. Adverse reactions to rifampicin in antituberculosis regimens. J. Antimicrob. Chemother. 1977, 3, 115–132. [Google Scholar] [CrossRef]
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.W.; Shen, Y.M.; Doi-Katayama, Y.; Tang, L.; Park, C.; Moore, B.S.; Hutchinson, C.R.; Floss, H.G. Direct evidence that the rifamycin polyketide synthase assembles polyketide chains processively. Proc. Natl. Acad. Sci. USA 1999, 96, 9051–9056. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wan, E.; Kim, C.J.; Floss, H.G.; Mahmud, T. Identification of tailoring genes involved in the modification of the polyketide backbone of rifamycin B by Amycolatopsis mediterranei S699. Microbiology 2005, 151, 2515–2528. [Google Scholar] [CrossRef] [Green Version]
- August, P.R.; Li, T.; Yoon, Y.J.; Ning, S.; Muller, R.; Yu, T.W.; Taylor, M.; Hoffmann, D.; Kim, C.G.; Zhang, X.; et al. Biosynthesis of the ansamycin antibiotic rifamycin: Deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei S699. Chem. Biol. 1998, 5, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Floss, H.G.; Yu, T.W. Rifamycins-mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- Tang, L.; Yoon, Y.J.; Choi, C.Y.; Hutchinson, C.R. Characterization of the enzymatic domains in the modular polyketide synthase involved in rifamycin B biosynthesis by Amycolatopsis mediterranei. Gene 1998, 216, 255–265. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Lessons from the rifamycin biosynthetic gene cluster. Curr. Opin. Chem. Biol. 1999, 3, 592–597. [Google Scholar] [CrossRef]
- Stratmann, A.; Toupet, C.; Schilling, W.; Traber, R.; Oberer, L.; Schupp, T. Intermediates of rifamycin polyketide synthase produced by an Amycolatopsis mediterranei mutant with inactivated rifF gene. Microbiology 1999, 145, 3365–3375. [Google Scholar] [CrossRef] [Green Version]
- Bierman, M.; Logan, R.; O’Brien, K.; Seno, E.T.; Rao, R.N.; Schoner, B.E. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 1992, 116, 43–49. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.M.; Tian, Y.Q.; Chiao, J.S.; Zhao, G.P.; Jiang, W.H. Stability of plasmid pA387 derivatives in Amylcolatopsis mediterranei producing rifamycin. Biotechnol. Lett. 2003, 25, 1647–1652. [Google Scholar] [CrossRef]
- Raahave, D. Paper disk-ager diffusion assay of penicillin in the presence of streptomycin. Antimicrob. Agents Chemother. 1974, 6, 603–605. [Google Scholar] [CrossRef] [Green Version]
- Arendrup, M.C.; Prakash, A.; Meletiadis, J.; Sharma, C.; Chowdhary, A. Comparison of EUCAST and CLSI reference microdilution MICs of eight antifungal compounds for Candida auris and associated tentative epidemiological cutoff values. Antimicrob. Agents Chemother. 2017, 61, e00485-17. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zhou, Q.; Ge, C.; Yang, J.; Li, H.; Chen, T.; Xie, H.; Cui, Y.; Shao, M.; Li, J.; et al. Rpn10 promotes tumor progression by regulating hypoxia-inducible factor 1 alpha through the PTEN/Akt signaling pathway in hepatocellular carcinoma. Cancer Lett. 2019, 447, 1–11. [Google Scholar] [CrossRef]
- Crump, J.A.; Luby, S.P.; Mintz, E.D. The global burden of typhoid fever. Bull. World Health Organ. 2004, 82, 346–353. [Google Scholar]
- Linington, R.G.; Robertson, M.; Gauthier, A.; Finlay, B.B.; van Soest, R.; Andersen, R.J. Caminoside A, an antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Organic Lett. 2002, 4, 4089–4092. [Google Scholar] [CrossRef]
- Sun, Y.H.; Rolan, H.G.; Tsolis, R.M. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J. Biol. Chem. 2007, 282, 33897–33901. [Google Scholar] [CrossRef] [Green Version]
- Lostroh, C.P.; Lee, C.A. The Salmonella pathogenicity island-1 type III secretion system. Microbes Infect. 2001, 3, 1281–1291. [Google Scholar] [CrossRef]
- Worrall, L.J.; Lameignere, E.; Strynadka, N.C. Structural overview of the bacterial injectisome. Curr. Opin. Microbiol. 2011, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, W.; Guo, Z.; Lu, C.; Shen, Y. Fusaric acid modulates type three secretion system of Salmonella enterica serovar Typhimurium. Biochem. Biophys. Res. Commun. 2014, 449, 455–459. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Song, R.; Guo, Z.; Wang, H.; Zhu, J.; Lu, C.; Shen, Y. Ansavaricins A–E: Five new streptovaricin derivatives from Streptomyces sp. S012. RSC Adv. 2017, 7, 5684–5693. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Li, X.; Li, J.; Yang, X.; Zhou, Y.; Lu, C.; Shen, Y. Licoflavonol is an inhibitor of the type three secretion system of Salmonella enterica serovar Typhimurium. Biochem. Biophys. Res. Commun. 2016, 477, 998–1004. [Google Scholar] [CrossRef]
- Curtiss, R.; Wanda, S.Y.; Gunn, B.M.; Zhang, X.; Tinge, S.A.; Ananthnarayan, V.; Mo, H.; Wang, S.; Kong, W. Salmonella enterica serovar Typhimurium strains with regulated delayed attenuation in vivo. Infect. Immun. 2009, 77, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lv, C.; Sun, W.; Li, Z.; Han, X.; Li, Y.; Shen, Y. Cytosporone B, an inhibitor of the type III secretion system of Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 2013, 57, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Wehrli, W.; Staeheli, M. The rifamycins-relation of chemical structure and action on RNA polymerase. Biochim. Biophys. Acta 1969, 182, 24–29. [Google Scholar] [CrossRef]
- Stratmann, A.; Schupp, T.; Toupet, C.; Schilling, W.; Oberer, L.; Traber, R. New insights into rifamycin B biosynthesis: Isolation of proansamycin B and 34a-deoxy-rifamycin was early macrocyclic intermediates indicating two separated biosynthetic pathways. J. Antibiot. 2002, 55, 396–406. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, J.; Tian, X.; Wu, X.; Li, T.; Lu, C.; Shen, Y. Isolation of 11,12- seco-rifamycin W derivatives reveals a cleavage pattern of the rifamycin ansa chain. Organic Lett. 2019, 21, 900–903. [Google Scholar] [CrossRef]
- Ghisalba, O.; Traxler, P.; Nuesch, J. Early intermediates in the biosynthesis of ansamycins. I. Isolation and identification of protorifamycin I. J. Antibiot. 1978, 31, 1124–1131. [Google Scholar] [CrossRef]
- Ghisalba, O.; Traxler, P.; Fuhrer, H.; Richter, W.J. Early intermediates in the biosynthesis of ansamycins. III. Isolation and identification of further 8-deoxyansamycins of the rifamycin-type. J. Antibiot. (Tokyo) 1980, 33, 847–856. [Google Scholar] [CrossRef] [Green Version]
- White, R.J.; Martinelli, E.; Lancini, G. Ansamycin biogenesis: Studies on a novel rifamycin isolated from a mutant strain of Nocardia mediterranei. Proc. Natl. Acad. Sci. USA 1974, 71, 3260–3264. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Frost, J.W. Kanosamine Biosynthesis: A Likely Source of the Aminoshikimate Pathway’s Nitrogen Atom. J. Am. Chem. Soc. 2002, 124, 10642–10643. [Google Scholar] [CrossRef]
- Arakawa, K.; Müller, R.; Mahmud, T.; Yu, T.W.; Floss, H.G. Characterization of the Early Stage Aminoshikimate Pathway in the Formation of 3-Amino-5-hydroxybenzoic Acid: The RifN Protein Specifically Converts Kanosamine into Kanosamine 6-Phosphate. J. Am. Chem. Soc. 2002, 124, 10644–10645. [Google Scholar] [CrossRef]
- Zhao, G.; Li, S.; Guo, Z.; Sun, M.; Lu, C. Overexpression of div8 increases the production and diversity of divergolides in Streptomyces sp. W112. RSC Adv. 2015, 5, 98209–98214. [Google Scholar] [CrossRef]
- Li, S.; Lu, C.; Ou, J.; Deng, J.; Shen, Y. Overexpression of hgc1 increases the production and diversity of hygrocins in Streptomyces sp. LZ35. RSC Adv. 2015, 5, 83843–83846. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Wang, H.; Lu, C. Pentaketide Ansamycin Microansamycins A-I from Micromonospora sp. Reveal Diverse Post-PKS Modifications. Organic Lett. 2018, 20, 1058–1061. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 b | 3 b | 4 b | 5 b | 6 b |
|---|---|---|---|---|---|---|
| 3 | 7.30, s | 7.67, s | 7.67, s | 7.67, s | 7.60, s | 7.61, s |
| 5 | 7.34, s | / | / | / | / | / |
| 8 | 7.68, s | 7.92, s | 7.93, s | 7.92, s | 7.96, s | 7.97, s |
| 13 | 1.74, s | 1.44, s | 2.17, s | 1.97, s | 2.10, d (1.0) | 2.04, d (1.0) |
| 14 | 2.29, s | 2.35, s | 2.35, s | 2.33, s | 2.36, s | 2.37, s |
| 17 | 6.23, d (10.7) | 6.47, d (11.2) | 6.47, d (11.0) | 6.52, d (11.3) | 6.24, d (10.8) | 6.28, d (11.1) |
| 18 | 6.64, t (13.1) | 6.80, dd (14.7, 11.2) | 6.80, dd (15.0, 11.2) | 7.13, dd (15.1, 11.3) | 6.49, dd (15.9, 11.0) | 6.53, dd (16.6, 11.2) |
| 19 | 5.93, dd (14.2, 6.7) | 6.03, dd (15.1, 7.9) | 6.04, dd (15.0, 8.0) | 6.07, dd (15.2, 10.2) | 6.08, dd (15.9, 6.7) | 6.02, dd (15.2, 6.4) |
| 20 | 2.24, m | 2.41, m | 2.41, m | 2.49, m | 2.31, m | 2.43, m |
| 21 | 3.63, d (8.0) | 3.76, m | 3.78, dd (8.9, 0.7) | 3.88, m | 4.03, m | 4.25, dd (8.6, 2.0) |
| 22 | 1.74, m | 1.88, m | 1.90, m | 1.65, m | 1.86, m | 1.94, m |
| 23 | 3.43, overlap | 3.50, dd (3.5, 8.7) | 3.50, dd (7.8, 4.6) | 3.27, dd (9.9, 3.1) | 3.47, dd (10.4, 1.9) | 3.46, dd (9.9, 2.3) |
| 24 | 1.67, m | 1.81, m | 1.84, m | 1.93, m | 1.78, m | 1.77, m |
| 25 | 3.82, d (8.8) | 3.74, m | 4.05, dd (11.3, 2.0) | 3.97, dd (8.6, 2.8) | 3.97, m | 3.95, dd (9.6, 0.7) |
| 26 | 1.56, m | 1.93, m | 2.01, m | 1.48, m | 1.38, m | 1.43, m |
| 27 | 3.71, d (5.84) | 3.98, d (3.6) | 3.46, t (8.5) | 3.17, t (10.2) | 4.30, m | 3.98, m |
| 28 | 2.53, overlap | 4.39, d (5.2) | 3.96, td (8.6, 3.4) | 2.51, m | 2.86, m | 2.61, m |
| 29 | 6.69, d (9.4) | 2.48, m 1.75, m | 2.76, dd (15.6, 3.5) 2.65, dd (15.6, 8.9) | 5.75, d (8.2, 0.7) | 6.26, dd (10.4, 1.1) | 6.42, dd (9.2, 2.0) |
| 30 | 2.04, s | 2.07, s | 2.07, s | 4.46, m 4.24, m | 2.07, s | 2.08, s |
| 31 | 0.89, d (6.1) | 0.99, d (6.8) | 0.99, d (6.2) | 1.14, d (6.9) | 0.91, d (6.9) | 3.53, d (4.7) |
| 32 | 0.78, d (6.4) | 1.02, d (7.0) | 0.97, d (5.8) | 0.66, d (7.0) | 1.05, d (7.0) | 1.08, d (3.2) |
| 33 | 0.82, d (7.2) | 0.85, d (7.0) | 0.95, d (6.6) | 1.06, d (7.0) | 0.71, d (6.8) | 0.74, d (6.8) |
| 34 | 0.66, d (6.4) | 0.83, d (7.0) | 1.04, d (6.5) | 0.80, d (6.4) | 0.38, d (7.0) | 0.37, d (7.0) |
| 34a | 0.85, d (6.9) | / | / | 5.12, d (4.1) | 4.02, m 4.00, m | 1.06, d (3.3) |
| 1′ | 4.82, s | 2.03, s | ||||
| 2′ | 4.09, s | |||||
| 3′ | 3.23, overlapped | |||||
| 4′ | 3.33, overlapped | |||||
| 5′ | 3.96, m | |||||
| 6′ | 1.19, d (5.8) |
| Position | 1 a | 2 b | 3 b | 4 b | 5 b | 6 b | 7 b | 8 b | 9 b | 10 b | 11 b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 130.1, C | 180.0, C | 181.3, C | 181.9, C | 181.1, C | 180.5, C | 181.0, C | 181.0, C | 181.0, C | 179.7, C | |
| 2 | 136.9, C | 142.0, C | 142.3, C | 143.1, C | 124.7, C | 141.2, C | 133.7, C | 142.0, C | 142.6, C | 141.8, C | |
| 3 | 104.5, CH | 117.4, CH | 117.9, CH | 116.7, CH | 118.9, CH | 118.7, CH | 118.2, CH | 118.5, CH | 119.0, CH | 119.2, CH | 119.0, CH |
| 4 | 147.7, C | 187.2, C | 188.0, C | 188.0, C | 188.0, C | ||||||
| 5 | 103.7, CH | 129.3, C | 129.1, C | ||||||||
| 6 | 153.4, C | 160.6, C | 160.3, C | 161.3, C | 164.0, C | 160.6, C | 160.0, C | 160.7, C | 160.7, C | 161.4, C | 162.0, C |
| 7 | 127.4, C | 132.5, C | 133.1, C | 133.5, C | 133.6, C | 133.1, C | 132.2, C | 132.3, C | 131.5, C | 132.9, C | |
| 8 | 123.1, CH | 131.2, CH | 132.0, CH | 132.1, CH | 132.5, CH | 132.0, CH | 131.6, CH | 132.0, CH | 132.4, CH | 132.0, CH | 131.2, C |
| 9 | 137.1, C | 124.5, C | 136.4, C | 126.9, C | 124.2, C | 129.1, C | |||||
| 10 | 122.6, C | 130.5, C | 130.7, C | 132.9, C | 132.0, C | 142.5, C | 132.5, C | 133.0, C | 133.0, C | ||
| 11 | 169.3, C | 200.2, C | 201.1, C | 201.3, C | 201.1, C | 201.2, C | 201.1, C | 201.4, C | 200.3, C | ||
| 12 | 125.9, C | 107.5, C | 211.0, C | 143.3, C | 142.6, C | 138.5, C | 140.7, C | 141.7, C | 142.2, C | 142.2, C | 142.9, C |
| 13 | 12.5, CH3 | 24.4, CH3 | 31.3, CH3 | 17.5, CH3 | 13.2, CH3 | 12.2, CH3 | 12.6, CH3 | 13.0, CH3 | 13.2, CH3 | 13.2, CH3 | 12.5, CH3 |
| 14 | 17.1, CH3 | 17.1, CH3 | 17.6, CH3 | 12.8, CH3 | 17.7, CH3 | 17.5, CH3 | 17.1, CH3 | 17.5, CH3 | 17.7, CH3 | 17.5, CH3 | 16.8, CH3 |
| 15 | 167.2, C | 170.2, C | 170.4, C | 170.8, C | 173.5, C | 173.3, C | 172.7, C | 170.9, C | 172.2, C | 173.3, C | 172.0, C |
| 16 | 121.0, C | 129.9, C | 130.1, C | 133.0, C | 132.5, C | 133.5, C | 133.4, C | 134.3, C | 133.4, C | 134.3, C | 132.9, C |
| 17 | 133.8, CH | 138.5, CH | 139.1, CH | 141.7, CH | 135.6, CH | 135.0, CH | 134.6, CH | 140.0, CH | 135.9, CH | 133.3, CH | 134.5, CH |
| 18 | 125.9, CH | 127.6, CH | 128.2, CH | 129.4, CH | 126.9, CH | 128.9, CH | 128.4, CH | 127.4, CH | 126.0, CH | 130.4, CH | 124.3, CH |
| 19 | 142.8, CH | 145.8, CH | 146.6, CH | 146.3, CH | 142.0, CH | 137.8, CH | 137.5, CH | 146.2, CH | 148.2, CH | 136.6, CH | 142.8, CH |
| 20 | 40.6, CH | 42.4, CH | 43.0, CH | 48.2, CH | 39.8, CH | 48.1, CH | 47.4, CH | 39.3, CH | 76.9, CH | 52.7, CH | 82.3, C |
| 21 | 73.2, CH | 75.7, CH | 76.3, CH | 78.0, CH | 75.5, CH | 72.2, CH | 71.7, CH | 76.6, CH | 76.6, CH | 73.9, CH | 86.4, CH |
| 22 | 36.2, CH | 35.8, CH | 37.3, CH | 43.8, CH | 35.0, CH | 35.5, CH | 35.2, CH | 34.6, CH | 35.2, CH | 50.0, CH | 48.3, CH |
| 23 | 76.7, CH | 78.1, CH | 79.6, CH | 82.1, CH | 79.5, CH | 79.4, CH | 79.1, CH | 79.5, CH | 80.7, CH | 211.3, C | 106.8, C |
| 24 | 35.0, CH | 37.6, CH | 39.1, CH | 34.5, CH | 38.7, CH | 38.7, CH | 38.5, CH | 38.4, CH | 38.9, CH | 50.8, CH | 43.0, CH |
| 25 | 69.9, CH | 75.4, CH | 84.2, CH | 73.7, CH | 71.9, CH | 71.6, CH | 71.2, CH | 71.7, CH | 72.2, CH | 71.5, CH | 73.8, CH |
| 26 | 38.2, CH | 34.5, CH | 44.8, CH | 41.8, CH | 44.5, CH | 44.1, CH | 44.0, CH | 44.5, CH | 44.5, CH | 42.8, CH | 43.7, CH |
| 27 | 72.3, CH | 89.2, CH | 83.4, CH | 74.2, CH | 69.2, CH | 74.5, CH | 68.8, CH | 69.6, CH | 69.7, CH | 68.9, CH | 68.3, CH |
| 28 | 37.1, CH | 71.2, CH | 81.5, CH | 49.8, CH | 46.8, CH | 41.4, CH | 49.7, CH | 50.3, CH | 50.3, CH | 49.7, CH | 48.9, CH |
| 29 | 147.0, CH | 46.6, CH2 | 49.1, CH2 | 145.8, CH | 140.5, CH | 147.0, CH | 141.9, CH | 142.0, CH | 142.5, CH | 141.3, CH | 142.0, CH |
| 30 | 20.6, CH3 | 20.6, CH3 | 21.1, CH3 | 65.7, CH2 | 20.9, CH3 | 20.6, CH3 | 20.1, CH3 | 65.4, CH2 | 20.9, CH3 | 20.9, CH3 | 20.3, CH3 |
| 31 | 16.8, CH3 | 17.2, CH3 | 17.9, CH3 | 20.3, CH3 | 18.7, CH3 | 63.9, CH2 | 63.5, CH2 | 18.2, CH3 | 26.7, CH3 | 65.1, CH2 | 28.7, CH3 |
| 32 | 10.1, CH3 | 11.3, CH3 | 11.6, CH3 | 12.2, CH3 | 11.8, CH3 | 12.5, CH3 | 12.0, CH3 | 11.3, CH3 | 14.5, CH3 | 15.5, CH3 | 13.0, CH3 |
| 33 | 10.4, CH3 | 10.3, CH3 | 11.3, CH3 | 12.1, CH3 | 9.5, CH3 | 9.3, CH3 | 8.8, CH3 | 9.4, CH3 | 9.7, CH3 | 8.5, CH3 | 8.6, CH3 |
| 34 | 9.0, CH3 | 12.6, CH3 | 14.9, CH3 | 12.9, CH3 | 12.3, CH3 | 11.5, CH3 | 11.7, CH3 | 11.9, CH3 | 12.3, CH3 | 12.2, CH3 | 12.4, CH3 |
| 34a | 16.9, CH3 | 94.8, CH | 66.4, CH2 | 20.0, CH3 | 64.3, CH2 | 65.0, CH2 | 65.0, CH2 | 65.0, CH2 | 64.8, CH2 | ||
| 1′ | 105.1, CH | 173.0, C | |||||||||
| 2′ | 70.8, CH | 21.5, CH3 | |||||||||
| 3′ | 73.0, CH | ||||||||||
| 4′ | 72.1, CH | ||||||||||
| 5′ | 71.1, CH | ||||||||||
| 6′ | 18.0, CH3 |
| Position | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| 3 | 7.61, s | 7.59, s | 7.58, s | 7.59, s | 7.43, s |
| 8 | 7.97, s | 7.93, s | 7.94, s | 7.92, s | 7.89, s |
| 13 | 2.09, s | 2.10, s | 2.08, s | 2.07, s | 2.04, s |
| 14 | 2.36, s | 2.38, s | 2.35, s | 2.34, s | 2.31, s |
| 17 | 6.29, dd (11.0, 1.0) | 6.51, br d (10.9) | 6.24, dd (10.8, 0.8) | 6.26, dd (10.9, 1.1) | 6.22, dd (11.4, 1.4) |
| 18 | 6.53, dd (15.9, 11.0) | 6.90, dd (16.1, 11.0) | 6.45, dd (15.9, 10.9) | 6.13, dd (15.1, 11.0) | 6.29, br d (14.4) |
| 19 | 6.01, dd (15.8, 7.0) | 6.36, dq (7.4, 1.3) | 5.95, br d (16.0) | 5.82, dd (15.1, 12.4) | 5.84, d (14.4) |
| 20 | 2.42, m | 2.32, m | 1.85, m | ||
| 21 | 4.26, dd (9.1, 1.8) | 4.05, br d (9.9) | 3.94, br s | 3.81, br d (10.0) | 3.80, d (10.5) |
| 22 | 1.95, m | 1.93, m | 2.00, m | 2.93, m | 1.79, m |
| 23 | 3.47, m | 3.49, dd (10.3, 1.8) | 3.42, dd (9.4, 2.6) | ||
| 24 | 1.78, m | 1.81, m | 1.71, m | 2.45, dd (7.4, 0.72) | 1.91, m |
| 25 | 3.97, dd (10.2, 1.0) | 3.97, dd (10.2, 1.2) | 3.92, dd (10.2, 1.0) | 3.86, br d (9.6) | 4.23, dd (10.2, 0.7) |
| 26 | 1.40, m | 1.42, m | 1.39, m | 1.32, m | 1.46, m |
| 27 | 4.37, m | 4.38, d (6.6) | 4.35, br s | 4.41, m | 4.28, br s |
| 28 | 2.66, m | 2.69, q (7.7) | 2.65, qd (7.9, 1.2) | 2.56, m | 2.56, m |
| 29 | 6.32, dd (9.4, 1.1) | 6.34, d (6.4) | 6.29, dd (9.5, 0.7) | 6.22, dd (9.2, 1.4) | 6.28, d (3.5) |
| 30 | 2.08, s | 4.36, br d (12.1) 4.23, br d (12.1) | 2.09, s | 2.04, s | 2.08, s |
| 31 | 3.54, m | 0.94, d (7.0) | 1.02, s | 4.37, d (12.8) 4.23, d (12.8) | 1.31, s |
| 32 | 1.08, d (7.0) | 1.06, d (7.0) | 1.17, d (7.0) | 1.03, d (6.8) | 1.13, d (6.5) |
| 33 | 0.73, d (6.8) | 0.74, d (6.8) | 0.74, d (6.8) | 1.13, d (7.4) | 0.97, d (7.2) |
| 34 | 0.39, d (7.0) | 0.42, d (7.0) | 0.40, d (7.0) | 0.44, d (7.0) | 0.55, d (8.1) |
| 34a | 3.56, m | 3.61, dd (10.9, 7.9) | 3.59, dd (10.9, 7.8) | 3.50, m 3.39, m | 3.46, m 3.35, m |
| 3.43, dd (10.9, 7.9) | 3.40, dd (10.9, 7.8) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, F.; Shi, Y.; Zhao, S.; Li, Z.; Wang, H.; Lu, C.; Shen, Y. 8-Deoxy-Rifamycin Derivatives from Amycolatopsis mediterranei S699 ΔrifT Strain. Biomolecules 2020, 10, 1265. https://doi.org/10.3390/biom10091265
Ye F, Shi Y, Zhao S, Li Z, Wang H, Lu C, Shen Y. 8-Deoxy-Rifamycin Derivatives from Amycolatopsis mediterranei S699 ΔrifT Strain. Biomolecules. 2020; 10(9):1265. https://doi.org/10.3390/biom10091265
Chicago/Turabian StyleYe, Feng, Yanrong Shi, Shengliang Zhao, Zhiying Li, Haoxin Wang, Chunhua Lu, and Yuemao Shen. 2020. "8-Deoxy-Rifamycin Derivatives from Amycolatopsis mediterranei S699 ΔrifT Strain" Biomolecules 10, no. 9: 1265. https://doi.org/10.3390/biom10091265
APA StyleYe, F., Shi, Y., Zhao, S., Li, Z., Wang, H., Lu, C., & Shen, Y. (2020). 8-Deoxy-Rifamycin Derivatives from Amycolatopsis mediterranei S699 ΔrifT Strain. Biomolecules, 10(9), 1265. https://doi.org/10.3390/biom10091265