
Quantitative Super-Resolution Imaging for the Analysis of GPCR Oligomerization

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Fixation and Immunofluorescence Staining for Antibody Validation
2.3. DNA—Antibody Coupling Reaction
2.4. Cell Fixation and Immunofluorescence Staining for DNA-PAINT Imaging
2.5. DNA-PAINT Imaging Experiments
2.6. DNA-PAINT Image Reconstruction and Cluster Analysis
2.7. qPAINT Analysis
2.8. Statistical Analysis
3. Results
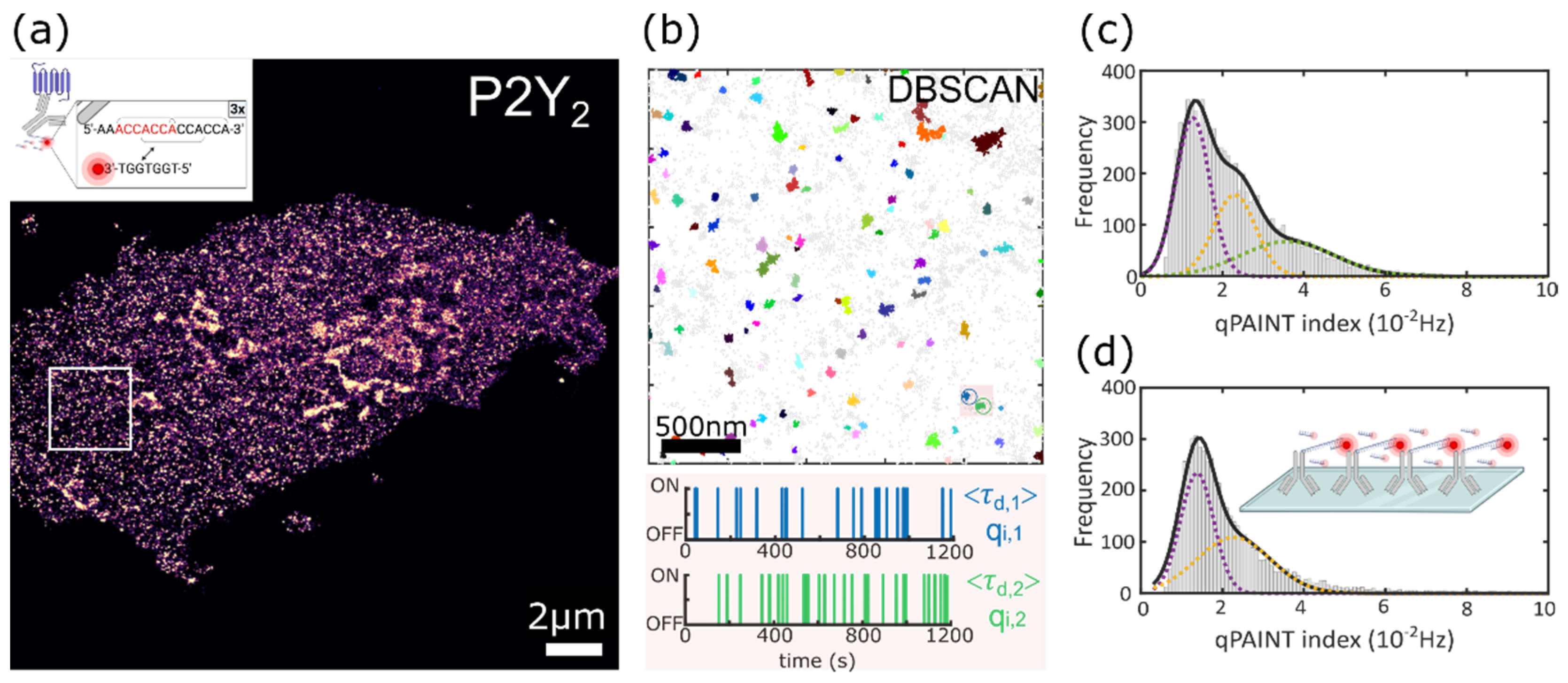
3.1. Super-Resolution Imaging of P2Y2 Receptors in AsPC-1 Cells Using DNA-PAINT
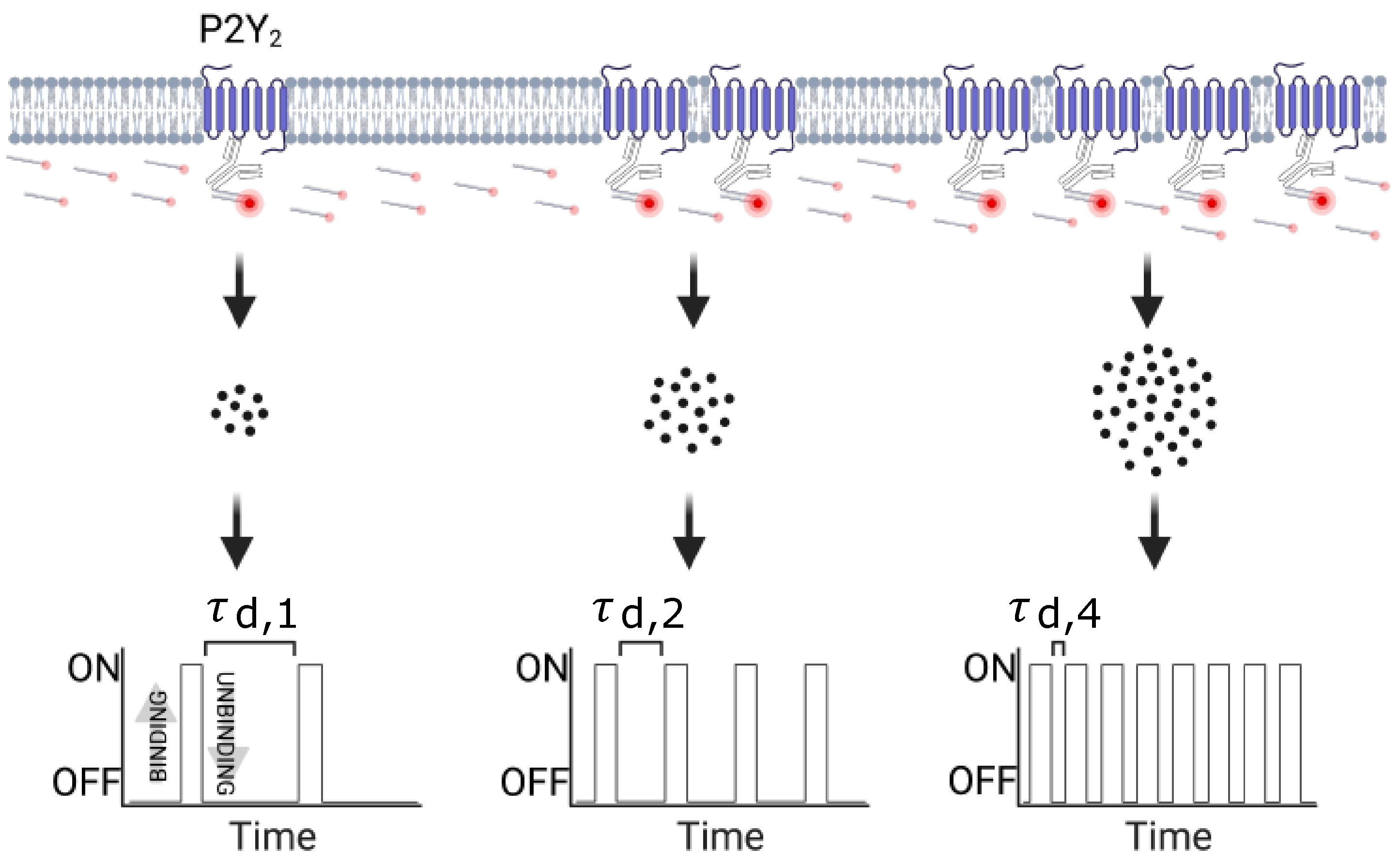
3.2. Quantitative Analysis of the P2Y2 Receptors Oligomerization via qPAINT Analysis
3.2.1. qPAINT Calibration
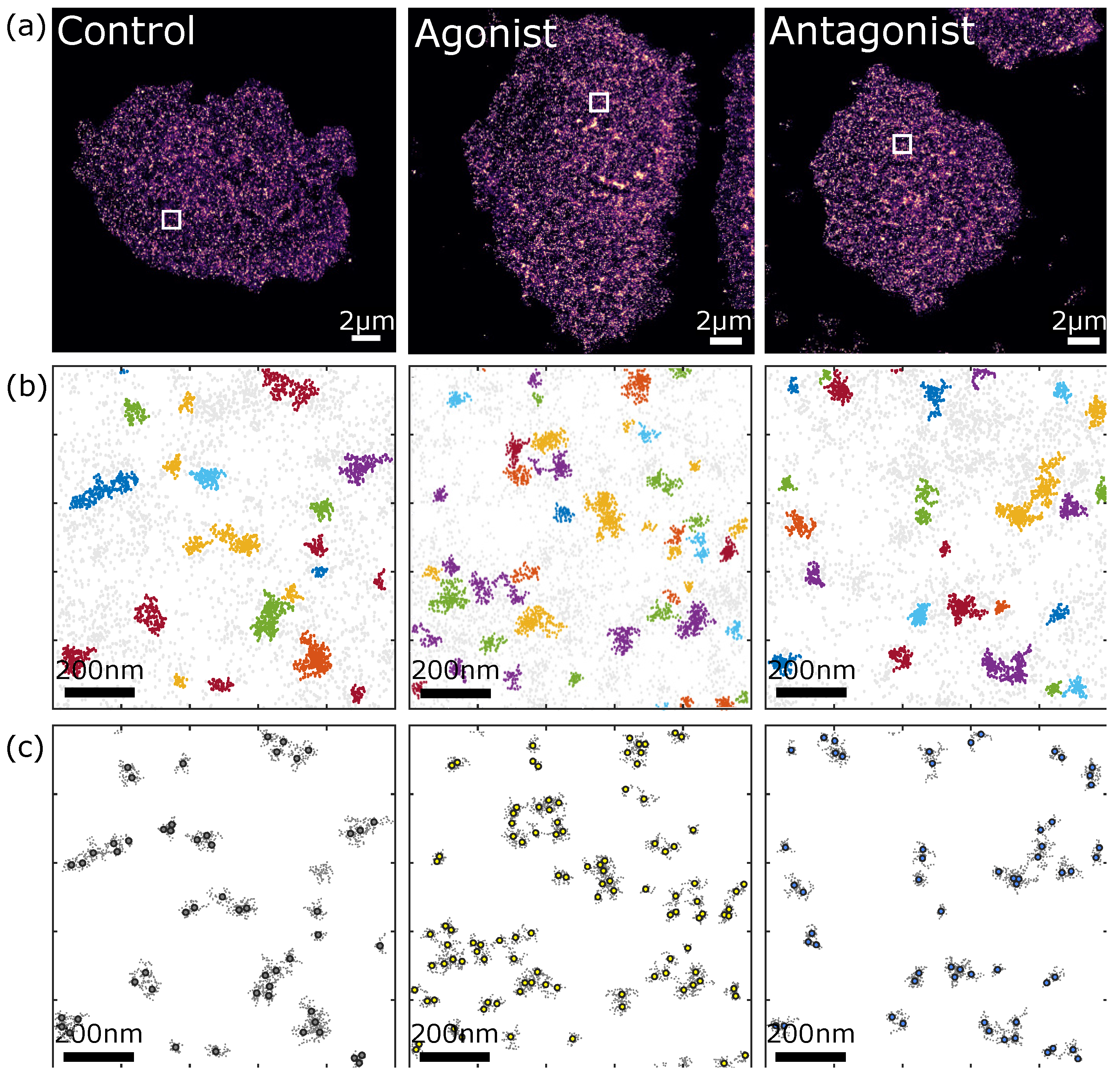
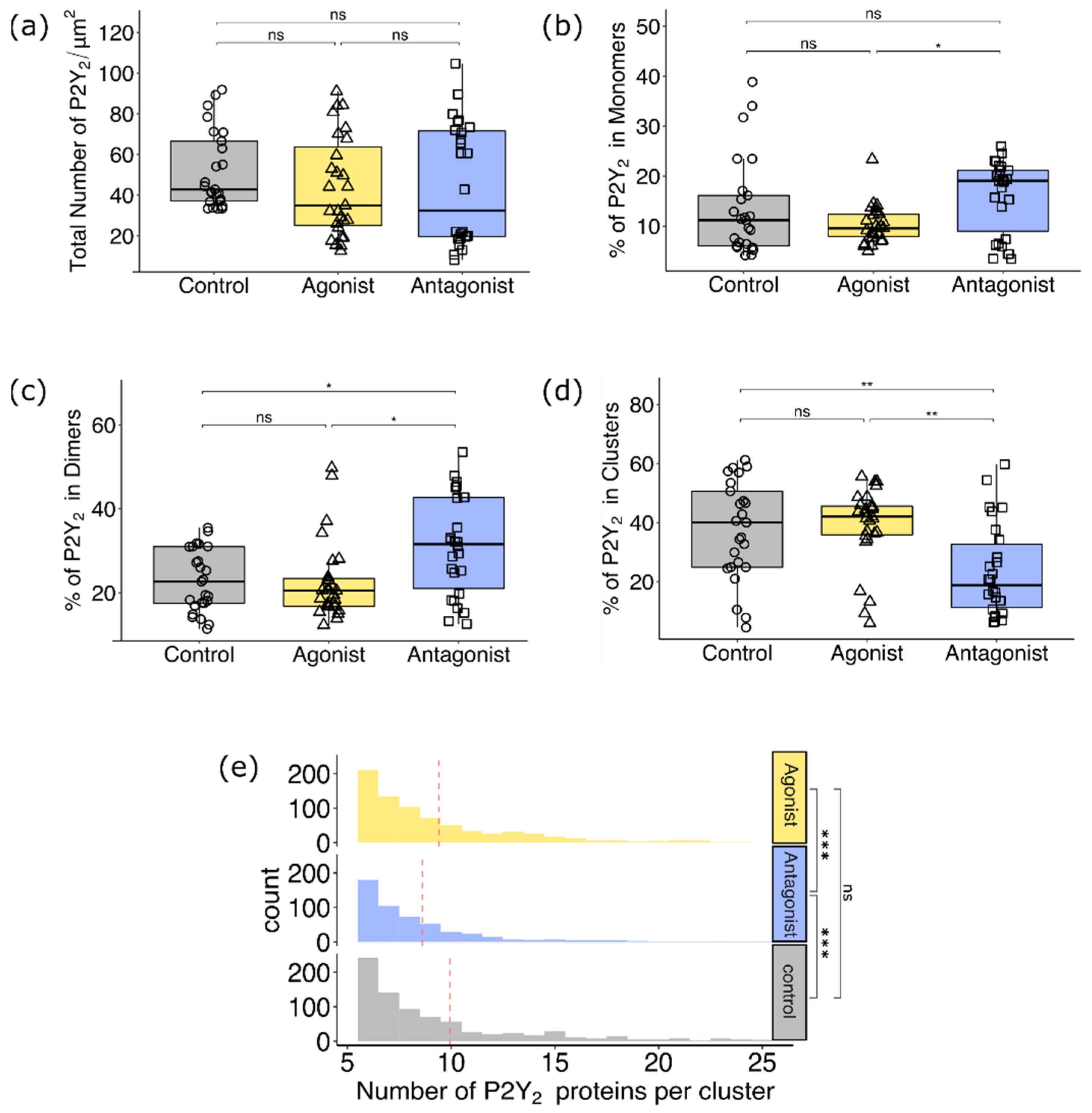
3.2.2. Comparison of P2Y2 Receptor Dimerization and Cluster Formation upon Agonist and Antagonist Treatment of AsPC-1 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wüthrich, K. The GPCR Network: A large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013, 12, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Israeli, H.; Degtjarik, O.; Fierro, F.; Chunilal, V.; Gill, A.K.; Roth, N.J.; Botta, J.; Prabahar, V.; Peleg, Y.; Chan, L.F.; et al. Structure reveals the activation mechanism of the MC4 receptor to initiate satiation signaling. Science 2021, 372, 808–814. [Google Scholar] [CrossRef]
- Jagannath, A.; Varga, N.; Dallmann, R.; Rando, G.; Gosselin, P.; Ebrahimjee, F.; Taylor, L.; Mosneagu, D.; Stefaniak, J.; Walsh, S.; et al. Adenosine integrates light and sleep signalling for the regulation of circadian timing in mice. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, R.; Canals, M. Experimental considerations for the assessment of in vivo and in vitro opioid pharmacology. Pharmacol. Ther. 2021, 107961. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Kumari, P.; Baidya, M.; Kise, R.; Cao, Y.; Dwivedi-Agnihotri, H.; Banerjee, R.; Li, X.X.; Cui, C.S.; Lee, J.D.; et al. Intrinsic bias at non-canonical, beta-arrestin-coupled seven transmembrane receptors. Mol. Cell 2021, 81, 1–17. [Google Scholar] [CrossRef]
- Stahl, E.L.; Bohn, L.M. Low Intrinsic Efficacy Alone Cannot Explain the Improved Side Effect Profiles of New Opioid Agonists. Biochemistry 2021. [Google Scholar] [CrossRef]
- Guo, X.; Li, Q.; Pi, S.; Xia, Y.; Mao, L. G protein-coupled purinergic P2Y receptor oligomerization: Pharmacological changes and dynamic regulation. Biochem. Pharmacol. 2021, 192, 114689. [Google Scholar] [CrossRef]
- Bouvier, M. Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2001, 2, 274–286. [Google Scholar] [CrossRef]
- Guo, H.; An, S.; Ward, R.; Yang, Y.; Liu, Y.; Guo, X.X.; Hao, Q.; Xu, T.R. Methods used to study the oligomeric structure of G-protein-coupled receptors. Biosci. Rep. 2017, 37, 37. [Google Scholar] [CrossRef] [Green Version]
- El Khamlichi, C.; Reverchon-Assadi, F.; Hervouet-Coste, N.; Blot, L.; Reiter, E.; Morisset-Lopez, S. Bioluminescence resonance energy transfer as a method to study protein-protein interactions: Application to G protein coupled receptor biology. Molecules 2019, 24, 537. [Google Scholar] [CrossRef] [Green Version]
- Işbilir, A.; Serfling, R.; Möller, J.; Thomas, R.; De Faveri, C.; Zabel, U.; Scarselli, M.; Beck-Sickinger, A.G.; Bock, A.; Coin, I.; et al. Determination of G-protein–coupled receptor oligomerization by molecular brightness analyses in single cells. Nat. Protoc. 2021, 16, 1419–1451. [Google Scholar] [CrossRef]
- Stoneman, M.R.; Biener, G.; Ward, R.J.; Pediani, J.D.; Badu, D.; Eis, A.; Popa, I.; Milligan, G.; Raicu, V. A general method to quantify ligand-driven oligomerization from fluorescence-based images. Nat. Methods 2019, 16, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Godin, A.G.; Costantino, S.; Lorenzo, L.E.; Swift, J.L.; Sergeev, M.; Ribeiro-da-Silva, A.; De Koninck, Y.; Wiseman, P.W. Revealing protein oligomerization and densities in situ using spatial intensity distribution analysis. Proc. Natl. Acad. Sci. USA 2011, 108, 7010–7015. [Google Scholar] [CrossRef] [Green Version]
- Sungkaworn, T.; Jobin, M.L.; Burnecki, K.; Weron, A.; Lohse, M.J.; Calebiro, D. Single-molecule imaging reveals receptor-G protein interactions at cell surface hot spots. Nature 2017, 550, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Rieken, F.; Wagner, J.; Sungkaworn, T.; Zabel, U.; Borzi, A.; Cocucci, E.; Zürn, A.; Lohse, M.J. Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. USA 2013, 110, 743–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarselli, M.; Annibale, P.; McCormick, P.J.; Kolachalam, S.; Aringhieri, S.; Radenovic, A.; Corsini, G.U.; Maggio, R. Revealing G-protein-coupled receptor oligomerization at the single-molecule level through a nanoscopic lens: Methods, dynamics and biological function. FEBS J. 2015, 283, 1197–1217. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.-P.; Zhang, X.-X.; Jiang, S.-H.; Tao, L.-Y.; Li, Q.; Zhu, L.-L.; Yang, M.-W.; Huo, Y.-M.; Jiang, Y.-S.; Tian, G.-A.; et al. Targeting Purinergic Receptor P2Y2 prevents the growth of pancreatic ductal adenocarcinoma by inhibiting cancer cell glycolysis. Clin. Cancer Res. 2019, 25, 1318–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef]
- Jungmann, R.; Avendaño, M.S.; Dai, M.; Woehrstein, J.B.; Agasti, S.S.; Feiger, Z.; Rodal, A.; Yin, P. Quantitative super-resolution imaging with qPAINT. Nat. Methods 2016, 13, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Guneri, D.; Hajjawi, M.O.R.; Shaw, K.; Patel, J.J.; Arnett, T.R. Activation of the P2Y2 receptor regulates bone cell function by enhancing ATP release. J. Endocrinol. 2017, 233, 341–356. [Google Scholar] [CrossRef]
- Séror, C.; Melki, M.T.; Subra, F.; Raza, S.Q.; Bras, M.; Saïdi, H.; Nardacci, R.; Voisin, L.; Paoletti, A.; Law, F.; et al. Extracellular ATP acts on P2Y2 purinergic receptors to facilitate HIV-1 infection. J. Exp. Med. 2011, 208, 1823–1834. [Google Scholar] [CrossRef]
- de la Rosa, G.; Gómez, A.I.; Baños, M.C.; Pelegrín, P. Signaling through purinergic receptor p2y2 enhances macrophage il-1β production. Int. J. Mol. Sci. 2020, 21, 4686. [Google Scholar] [CrossRef]
- Li, W.; Qiu, Y.; Zhang, H.; Tian, X.; Fang, W. P2Y2 Receptor and EGFR Cooperate to Promote Prostate Cancer Cell Invasion via. PLoS ONE 2015, 10, e0133165. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Yang, H.; Zhang, H.; Tian, X. ATP-P2Y2- b -catenin axis promotes cell invasion in breast cancer cells. Cancer Sci. 2017, 108, 1318–1327. [Google Scholar] [CrossRef]
- Woods, L.T.; Jasmer, K.J.; Muñoz Forti, K.; Shanbhag, V.C.; Camden, J.M.; Erb, L.; Petris, M.J.; Weisman, G.A. P2Y2 receptors mediate nucleotide-induced EGFR phosphorylation and stimulate proliferation and tumorigenesis of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2020, 109, 104808. [Google Scholar] [CrossRef]
- Abe, M.; Watanabe, K.; Kuroda, Y.; Nakagawa, T.; Higashi, H. Homodimer formation by the ATP/UTP receptor P2Y2 via disulfide bridges. J. Biochem. 2018, 163, 475–480. [Google Scholar] [CrossRef]
- Kotevic, I.; Kirschner, K.M.; Porzig, H.; Baltensperger, K. Constitutive interaction of the P2Y2 receptor with the hematopoietic cell-specific G protein Gα16 and evidence for receptor oligomers. Cell. Signal. 2005, 17, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Shimano, T.; Chu, T.M. Differential localization of human pancreas cancer-associated antigen and carcinoembryonic antigen in homologous pancreatic tumoral xenograft. J. Natl. Cancer Inst. 1981, 67, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.M.; Mirza, T.A.; Feakins, R.M.; Seedhar, A.; Elia, G.; Hart, I.R.; Kocher, H.M. Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, β-catenin, and ezrin expression in tumor cells. Am. J. Pathol. 2009, 175, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Choi, R.C.Y.; Chu, G.K.Y.; Siow, N.L.; Yung, A.W.Y.; Yung, L.Y.; Lee, P.S.C.; Lo, C.C.W.; Simon, J.; Dong, T.T.X.; Barnard, E.A.; et al. Activation of utp-sensitive p2y2 receptor induces the expression of cholinergic genes in cultured cortical neurons: A signaling cascade triggered by ca2+ mobilization and extracellular regulated kinase phosphorylations. Mol. Pharmacol. 2013, 84, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoncelli, S.; Griffié, J.; Williamson, D.J.; Bibby, J.; Bray, C.; Zamoyska, R.; Cope, A.P.; Owen, D.M. Multi-color Molecular Visualization of Signaling Proteins Reveals How C-Terminal Src Kinase Nanoclusters Regulate T Cell Receptor Activation. Cell Rep. 2020, 33, 108523. [Google Scholar] [CrossRef] [PubMed]
- Pengo, T.; Holden, S.J.; Manley, S. PALMsiever: A tool to turn raw data into results for single-molecule localization microscopy. Bioinformatics 2015, 31, 797–798. [Google Scholar] [CrossRef] [Green Version]
- MATLAB. Version 9.9.0 (R2021a); The MathWorks Inc.: Natick, MA, USA, 2021. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; The R Project for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Allaire, J. RStudio: Integrated Development Environment for R; The R Project for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Sposini, S.; Hanyaloglu, A.C. Spatial encryption of G protein-coupled receptor signaling in endosomes; Mechanisms and applications. Biochem. Pharmacol. 2017, 143, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Salmerón, C.; Wiley, S.Z.; Insel, P.A. GPCRs in pancreatic adenocarcinoma: Contributors to tumour biology and novel therapeutic targets. Br. J. Pharmacol. 2020, 177, 2434–2455. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.; Jungmann, R. Up to 100-fold speed-up and multiplexing in optimized DNA-PAINT. Nat. Methods 2020, 17, 789–791. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996. [Google Scholar]
- Jayasinghe, I.; Clowsley, A.H.; Lin, R.; Lutz, T.; Harrison, C.; Green, E.; Baddeley, D.; Di Michele, L.; Soeller, C. True Molecular Scale Visualization of Variable Clustering Properties of Ryanodine Receptors. Cell Rep. 2018, 22, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.H.; Liu, M.; Nolting, B.; Go, J.G.; Gervay-Hague, J.; Liu, G.Y. A nanoengineering approach for investigation and regulation of protein immobilization. ACS Nano 2008, 2, 2374–2384. [Google Scholar] [CrossRef] [Green Version]
- Kasai, R.S.; Suzuki, K.G.N.; Prossnitz, E.R.; Koyama-Honda, I.; Nakada, C.; Fujiwara, T.K.; Kusumi, A. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 2011, 192, 463–480. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.J.; Pediani, J.D.; Godin, A.G.; Milligan, G. Regulation of oligomeric organization of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor observed by spatial intensity distribution analysis. J. Biol. Chem. 2015, 290, 12844–12857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, W.B.; Geggier, P.; Holsey, M.D.; Gilmore, G.T.; Pati, A.K.; Meszaros, J.; Terry, D.S.; Mathiasen, S.; Kaliszewski, M.J.; McCauley, M.D.; et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nat. Methods 2021, 18, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Siddig, S.; Aufmkolk, S.; Doose, S.; Jobin, M.L.; Werner, C.; Sauer, M.; Calebiro, D. Super-resolution imaging reveals the nanoscale organization of metabotropic glutamate receptors at presynaptic active zones. Sci. Adv. 2020, 6, eaay7193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, K.C.; Fanelli, F.; Huhtaniemi, I.T.; Hanyaloglu, A.C. Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J. Biol. Chem. 2015, 290, 3875–3892. [Google Scholar] [CrossRef] [Green Version]
- Fischer, L.S.; Klingner, C.; Schlichthaerle, T.; Strauss, M.T.; Böttcher, R.; Fässler, R.; Jungmann, R.; Grashoff, C. Quantitative single-protein imaging reveals molecular complex formation of integrin, talin, and kindlin during cell adhesion. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Marsango, S.; Caltabiano, G.; Jiménez-Rosés, M.; Millan, M.J.; Pediani, J.D.; Ward, R.J.; Milligan, G. A molecular basis for selective antagonist destabilization of dopamine D3 receptor quaternary organization. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.J.; Pediani, J.D.; Marsango, S.; Jolly, R.; Stoneman, M.R.; Biener, G.; Handel, T.M.; Raicu, V.; Milligan, G. Chemokine receptor CXCR4 oligomerization is disrupted selectively by the antagonist ligand IT1t. J. Biol. Chem. 2021, 296, 100139. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, M.D.; Tomas Bort, E.; Grose, R.P.; McCormick, P.J.; Simoncelli, S. Quantitative Super-Resolution Imaging for the Analysis of GPCR Oligomerization. Biomolecules 2021, 11, 1503. https://doi.org/10.3390/biom11101503
Joseph MD, Tomas Bort E, Grose RP, McCormick PJ, Simoncelli S. Quantitative Super-Resolution Imaging for the Analysis of GPCR Oligomerization. Biomolecules. 2021; 11(10):1503. https://doi.org/10.3390/biom11101503
Chicago/Turabian StyleJoseph, Megan D., Elena Tomas Bort, Richard P. Grose, Peter J. McCormick, and Sabrina Simoncelli. 2021. "Quantitative Super-Resolution Imaging for the Analysis of GPCR Oligomerization" Biomolecules 11, no. 10: 1503. https://doi.org/10.3390/biom11101503
APA StyleJoseph, M. D., Tomas Bort, E., Grose, R. P., McCormick, P. J., & Simoncelli, S. (2021). Quantitative Super-Resolution Imaging for the Analysis of GPCR Oligomerization. Biomolecules, 11(10), 1503. https://doi.org/10.3390/biom11101503