
Gut Metabolite Trimethylamine N-Oxide Protects INS-1 β-Cell and Rat Islet Function under Diabetic Glucolipotoxic Conditions

 , , ,
, , , 
Abstract
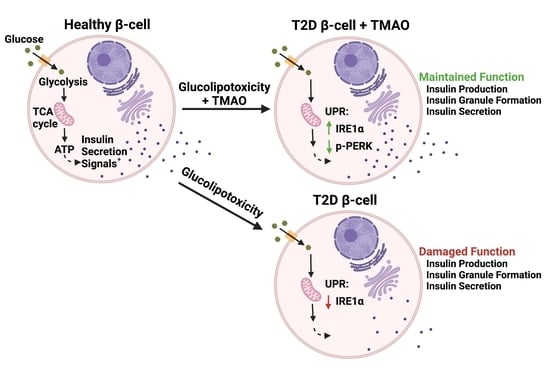
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell and Tissue Culture
2.2. Experimental Culture Treatments
2.3. Tetrazolium Salt MTT Viability Assay
2.4. Annexin V (AV) and 7-Aminoactinomycin D (7-AAD) Survival Assay
2.5. [3H]-Thymidine Incorporation Proliferation Assay
2.6. Glucose-Simulated Insulin Secretion (GSIS) Assay
2.7. Scanning Transmission Electron Microscopy (STEM)
2.8. Glutathione (GSH) Assay
2.9. qPCR
2.10. Western Blots
2.11. Statistical Analysis
3. Results
3.1. TMAO Does Not Alter GLT-Mediated Reduction of β-Cell Mass
3.2. TMAO Normalizes GLT-Damaged β-Cell and Islet Function
3.3. TMAO Recovers the Decrease in Insulin Granule Number Induced by GLT
3.4. TMAO Does Not Alter GLT-Mediated Changes to GSH or Redox Potential
3.5. TMAO Normalizes GLT Mediated Changes to Unfolded Protein Response (UPR) Components
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kones, R.; Rumana, U. Cardiometabolic diseases of civilization: History and maturation of an evolving global threat. An update and call to action. Ann. Med. 2017, 49, 260–274. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Goran, M.I.; Bosy-Westphal, A.; King, J.C.; Schmidt, L.A.; Schwarz, J.M.; Stice, E.; Sylvetsky, A.C.; Turnbaugh, P.J.; Bray, G.A.; et al. Pathways and mechanisms linking dietary components to cardiometabolic disease: Thinking beyond calories. Obes. Rev. 2018, 19, 1205–1235. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet and Lifestyle Drive the Pandemic of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Z.; Sun, T.; Huang, H.; Chen, S.; Chen, L.; Luo, C.; Yang, W.; Yang, X.; Yao, P.; Cheng, J.; et al. Association between microbiota-dependent metabolite trimethylamine-N-oxide and type 2 diabetes. Am. J. Clin. Nutr. 2017, 106, 888–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svingen, G.F.; Schartum-Hansen, H.; Pedersen, E.R.; Ueland, P.M.; Tell, G.S.; Mellgren, G.; Njolstad, P.R.; Seifert, R.; Strand, E.; Karlsson, T.; et al. Prospective Associations of Systemic and Urinary Choline Metabolites with Incident Type 2 Diabetes. Clin. Chem. 2016, 62, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Awwad, H.M.; Rabagny, Y.; Graeber, S.; Herrmann, W.; Geisel, J. Plasma trimethylamine N-oxide concentration is associated with choline, phospholipids, and methyl metabolism. Am. J. Clin. Nutr. 2016, 103, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.J.; Lipworth, L.P.; Shu, X.O.; Blot, W.J.; Xiang, Y.B.; Steinwandel, M.D.; Li, H.; Gao, Y.T.; Zheng, W.; Yu, D. Associations of choline-related nutrients with cardiometabolic and all-cause mortality: Results from 3 prospective cohort studies of blacks, whites, and Chinese. Am. J. Clin. Nutr. 2020, 111, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Argyridou, S.; Davies, M.J.; Biddle, G.J.H.; Bernieh, D.; Suzuki, T.; Dawkins, N.P.; Rowlands, A.V.; Khunti, K.; Smith, A.C.; Yates, T. Evaluation of an 8-Week Vegan Diet on Plasma Trimethylamine-N-Oxide and Postchallenge Glucose in Adults with Dysglycemia or Obesity. J. Nutr. 2021, 151, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar]
- Rebouche, C.J.; Chenard, C.A. Metabolic fate of dietary carnitine in human adults: Identification and quantification of urinary and fecal metabolites. J. Nutr. 1991, 121, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Al-Waiz, M.; Mitchell, S.C.; Idle, J.R.; Smith, R.L. The metabolism of 14C-labelled trimethylamine and its N-oxide in man. Xenobiotica 1987, 17, 551–558. [Google Scholar] [CrossRef]
- Ufnal, M.; Zadlo, A.; Ostaszewski, R. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef]
- Mitchell, S.M.; Milan, A.M.; Mitchell, C.J.; Gillies, N.A.; D’Souza, R.F.; Zeng, N.; Ramzan, F.; Sharma, P.; Knowles, S.O.; Roy, N.C.; et al. Protein Intake at Twice the RDA in Older Men Increases Circulatory Concentrations of the Microbiome Metabolite Trimethylamine-N-Oxide (TMAO). Nutrients 2019, 11, 2207. [Google Scholar] [CrossRef] [Green Version]
- Martinez-del Campo, A.; Bodea, S.; Hamer, H.A.; Marks, J.A.; Haiser, H.J.; Turnbaugh, P.J.; Balskus, E.P. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 2015, 6, e00042-15. [Google Scholar] [CrossRef] [Green Version]
- Krueger, S.K.; Williams, D.E. Mammalian flavin-containing monooxygenases: Structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 2005, 106, 357–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canyelles, M.; Tondo, M.; Cedo, L.; Farras, M.; Escola-Gil, J.C.; Blanco-Vaca, F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 2018, 19, 3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Richey, J.J.; Zigler, M.C.; Cuevas, L.M.; Gonzalez, A.; Vazquez-Baeza, Y.; Battson, M.L.; Smithson, A.T.; Gilley, A.D.; et al. Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. J. Physiol. 2019, 597, 2361–2378. [Google Scholar] [CrossRef] [Green Version]
- Krueger, E.S.; Lloyd, T.S.; Tessem, J.S. The Accumulation and Molecular Effects of Trimethylamine N-Oxide on Metabolic Tissues: It’s Not All Bad. Nutrients 2021, 13, 2873. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart. Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef]
- Lever, M.; George, P.M.; Slow, S.; Bellamy, D.; Young, J.M.; Ho, M.; McEntyre, C.J.; Elmslie, J.L.; Atkinson, W.; Molyneux, S.L.; et al. Betaine and Trimethylamine-N-Oxide as Predictors of Cardiovascular Outcomes Show Different Patterns in Diabetes Mellitus: An Observational Study. PLoS ONE 2014, 9, e114969. [Google Scholar] [CrossRef] [Green Version]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; von Eckardstein, A. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Latkovskis, G.; Kuka, J.; Strele, I.; Konrade, I.; Grinberga, S.; Hartmane, D.; Pugovics, O.; Erglis, A.; Liepinsh, E. Diabetes is Associated with Higher Trimethylamine N-oxide Plasma Levels. Exp. Clin. Endocrinol. Diabetes 2016, 124, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Ling, A.V.; Manthena, P.V.; Gearing, M.E.; Graham, M.J.; Crooke, R.M.; Croce, K.J.; Esquejo, R.M.; Clish, C.B.; Vicent, D.; et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015, 6, 6498. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.T.; Manson, A.C.; DeLyser, M.R.; Noid, W.G.; Cremer, P.S. Trimethylamine N-oxide stabilizes proteins via a distinct mechanism compared with betaine and glycine. Proc. Natl. Acad. Sci. USA 2017, 114, 2479–2484. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.H.; Xin, F.Z.; Zhou, D.; Xue, Y.Q.; Liu, X.L.; Yang, R.X.; Pan, Q.; Fan, J.G. Trimethylamine N-oxide attenuates high-fat high-cholesterol diet-induced steatohepatitis by reducing hepatic cholesterol overload in rats. World J. Gastroenterol. 2019, 25, 2450–2462. [Google Scholar] [CrossRef]
- Mondal, J.; Stirnemann, G.; Berne, B.J. When does trimethylamine N-oxide fold a polymer chain and urea unfold it? J. Phys. Chem. B 2013, 117, 8723–8732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.L.; Chuang, D.T. Natural osmolyte trimethylamine N-oxide corrects assembly defects of mutant branched-chain α-ketoacid decarboxylase in maple syrup urine disease. J. Biol. Chem. 2001, 276, 40241–40246. [Google Scholar] [CrossRef] [Green Version]
- Sackett, D.L. Natural osmolyte trimethylamine N-oxide stimulates tubulin polymerization and reverses urea inhibition. Am. J. Physiol. 1997, 273, R669–R676. [Google Scholar] [CrossRef]
- Villalobos, A.R.; Renfro, J.L. Trimethylamine oxide suppresses stress-induced alteration of organic anion transport in choroid plexus. J. Exp. Biol. 2007, 210, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Bolen, D.W. A naturally occurring protective system in urea-rich cells: Mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry 1997, 36, 9101–9108. [Google Scholar] [CrossRef]
- Steiner, D.J.; Kim, A.; Miller, K.; Hara, M. Pancreatic islet plasticity: Interspecies comparison of islet architecture and composition. Islets 2010, 2, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Misawa, R.; Zielinski, M.C.; Cowen, P.; Jo, J.; Periwal, V.; Ricordi, C.; Khan, A.; Szust, J.; Shen, J.; et al. Regional differences in islet distribution in the human pancreas—Preferential beta-cell loss in the head region in patients with type 2 diabetes. PLoS ONE 2013, 8, e67454. [Google Scholar] [CrossRef]
- Tiedge, M.; Lenzen, S. Regulation of glucokinase and GLUT-2 glucose-transporter gene expression in pancreatic B-cells. Biochem. J. 1991, 279 Pt 3, 899–901. [Google Scholar] [CrossRef] [Green Version]
- Schuit, F.C.; De Vos, A.; Moens, K.; Quartier, E.; Heimberg, H. Glucose-induced B-cell recruitment and the expression of hexokinase isoenzymes. Adv. Exp. Med. Biol. 1997, 426, 259–266. [Google Scholar] [CrossRef]
- Boland, B.B.; Rhodes, C.J.; Grimsby, J.S. The dynamic plasticity of insulin production in β-cells. Mol. Metab. 2017, 6, 958–973. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Welsh, N.; Margulis, B.; Borg, L.A.; Wiklund, H.J.; Saldeen, J.; Flodstrom, M.; Mello, M.A.; Andersson, A.; Pipeleers, D.G.; Hellerstrom, C.; et al. Differences in the expression of heat-shock proteins and antioxidant enzymes between human and rodent pancreatic islets: Implications for the pathogenesis of insulin-dependent diabetes mellitus. Mol. Med. 1995, 1, 806–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, D.F.; Cunningham, D.; Spigelman, L.; Aten, B. Insulin biosynthesis: Evidence for a precursor. Science 1967, 157, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F.; Terris, S.; Chan, S.J.; Rubenstein, A.H. Chemical and biological aspects of insulin and proinsulin. Acta Med. Scand. Suppl. 1976, 601, 55–107. [Google Scholar] [PubMed]
- Kanda, Y.; Hashiramoto, M.; Shimoda, M.; Hamamoto, S.; Tawaramoto, K.; Kimura, T.; Hirukawa, H.; Nakashima, K.; Kaku, K. Dietary restriction preserves the mass and function of pancreatic β cells via cell kinetic regulation and suppression of oxidative/ER stress in diabetic mice. J. Nutr. Biochem. 2015, 26, 219–226. [Google Scholar] [CrossRef]
- Weir, G.C.; Gaglia, J.; Bonner-Weir, S. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 249–256. [Google Scholar] [CrossRef]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Jensen, D.M.; Hendricks, K.V.; Mason, A.T.; Tessem, J.S. Good Cop, Bad Cop: The Opposing Effects of Macrophage Activation State on Maintaining or Damaging Functional β-Cell Mass. Metabolites 2020, 10, 485. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and β-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Gregg, B.; Blandino-Rosano, M.; Cras-Meneur, C.; Bernal-Mizrachi, E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol. Asp. Med. 2015, 42, 19–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudish, L.I.; Reusch, J.E.; Sussel, L. β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Investig. 2019, 129, 4001–4008. [Google Scholar] [CrossRef] [Green Version]
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; et al. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 2019, 28, 759–772.e10. [Google Scholar] [CrossRef]
- Bitner, B.F.; Ray, J.D.; Kener, K.B.; Herring, J.A.; Tueller, J.A.; Johnson, D.K.; Tellez Freitas, C.M.; Fausnacht, D.W.; Allen, M.E.; Thomson, A.H.; et al. Common gut microbial metabolites of dietary flavonoids exert potent protective activities in β-cells and skeletal muscle cells. J. Nutr. Biochem. 2018, 62, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessem, J.S.; Moss, L.G.; Chao, L.C.; Arlotto, M.; Lu, D.; Jensen, M.V.; Stephens, S.B.; Tontonoz, P.; Hohmeier, H.E.; Newgard, C.B. Nkx6.1 regulates islet β-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 5242–5247. [Google Scholar] [CrossRef] [Green Version]
- Schisler, J.C.; Fueger, P.T.; Babu, D.A.; Hohmeier, H.E.; Tessem, J.S.; Lu, D.; Becker, T.C.; Naziruddin, B.; Levy, M.; Mirmira, R.G.; et al. Stimulation of human and rat islet β-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Mol. Cell. Biol. 2008, 28, 3465–3476. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.H.; Crider, B.P.; Mccorkle, K.; Alford, M.; Unger, R.H. Inhibition of Glucose-Transport into Rat Islet Cells by Immunoglobulins from Patients with New-Onset Insulin-Dependent Diabetes-Mellitus. N. Engl. J. Med. 1990, 322, 653–659. [Google Scholar] [CrossRef]
- Veluthakal, R.; Arora, D.K.; Goalstone, M.L.; Kowluru, R.A.; Kowluru, A. Metabolic Stress Induces Caspase-3 Mediated Degradation and Inactivation of Farnesyl and Geranylgeranyl Transferase Activities in Pancreatic β-Cells. Cell. Physiol. Biochem. 2016, 39, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Suresh, M.V.; Kowluru, A. Down-regulation of expression and function of nucleoside diphosphate kinase in insulin-secreting β-cells under in vitro conditions of glucolipotoxicity. Mol. Cell. Biochem. 2009, 329, 121–129. [Google Scholar] [CrossRef]
- Kornelius, E.; Li, H.H.; Peng, C.H.; Yang, Y.S.; Chen, W.J.; Chang, Y.Z.; Bai, Y.C.; Liu, S.; Huang, C.N.; Lin, C.L. Liraglutide protects against glucolipotoxicity-induced RIN-m5F β-cell apoptosis through restoration of PDX1 expression. J. Cell. Mol. Med. 2019, 23, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Joly, E.; El-Assaad, W.; Roduit, R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: Role in β-cell adaptation and failure in the etiology of diabetes. Diabetes 2002, 51 (Suppl. 3), S405–S413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsabeeh, N.; Chausse, B.; Kakimoto, P.A.; Kowaltowski, A.J.; Shirihai, O. Cell culture models of fatty acid overload: Problems and solutions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 143–151. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Cunha, D.A.; Ladriere, L.; Igoillo-Esteve, M.; Bugliani, M.; Marchetti, P.; Cnop, M. In vitro use of free fatty acids bound to albumin: A comparison of protocols. Biotechniques 2015, 58, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Paolisso, G.; Tataranni, P.A.; Foley, J.E.; Bogardus, C.; Howard, B.V.; Ravussin, E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia 1995, 38, 1213–1217. [Google Scholar] [CrossRef]
- Garcia, E.; Wolak-Dinsmore, J.; Wang, Z.; Li, X.S.; Bennett, D.W.; Connelly, M.A.; Otvos, J.D.; Hazen, S.L.; Jeyarajah, E.J. NMR quantification of trimethylamine-N-oxide in human serum and plasma in the clinical laboratory setting. Clin. Biochem. 2017, 50, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Prokopienko, A.J.; West, R.E., 3rd; Schrum, D.P.; Stubbs, J.R.; Leblond, F.A.; Pichette, V.; Nolin, T.D. Metabolic Activation of Flavin Monooxygenase-mediated Trimethylamine-N-Oxide Formation in Experimental Kidney Disease. Sci. Rep. 2019, 9, 15901. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.; Prokopienko, A.J.; West, R.E., 3rd; Nolin, T.D.; Stubbs, J.R. Decreased Kidney Function Is Associated with Enhanced Hepatic Flavin Monooxygenase Activity and Increased Circulating Trimethylamine N-Oxide Concentrations in Mice. Drug Metab. Dispos. 2018, 46, 1304–1309. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Buffa, J.A.; Roberts, A.B.; Sangwan, N.; Skye, S.M.; Li, L.; Ho, K.J.; Varga, J.; DiDonato, J.A.; Tang, W.H.W.; et al. Targeted Inhibition of Gut Microbial Trimethylamine N-Oxide Production Reduces Renal Tubulointerstitial Fibrosis and Functional Impairment in a Murine Model of Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1239–1255. [Google Scholar] [CrossRef]
- Al-Obaide, M.A.I.; Singh, R.; Datta, P.; Rewers-Felkins, K.A.; Salguero, M.V.; Al-Obaidi, I.; Kottapalli, K.R.; Vasylyeva, T.L. Gut Microbiota-Dependent Trimethylamine-N-oxide and Serum Biomarkers in Patients with T2DM and Advanced CKD. J. Clin. Med. 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.Y.; Xia, G.H.; Lu, J.Q.; Chen, M.X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Troseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjorndal, B.; Halvorsen, B.; et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Shih, D.M.; Warrier, M.; Helsley, R.N.; Burrows, A.; Ferguson, D.; Brown, A.L.; Gromovsky, A.D.; Heine, M.; Chatterjee, A.; et al. The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep. 2017, 19, 2451–2461. [Google Scholar] [CrossRef] [Green Version]
- McEntyre, C.J.; Lever, M.; Chambers, S.T.; George, P.M.; Slow, S.; Elmslie, J.L.; Florkowski, C.M.; Lunt, H.; Krebs, J.D. Variation of betaine, N,N-dimethylglycine, choline, glycerophosphorylcholine, taurine and trimethylamine-N-oxide in the plasma and urine of overweight people with type 2 diabetes over a two-year period. Ann. Clin. Biochem. 2015, 52, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Griffin, L.E.; Djuric, Z.; Angiletta, C.J.; Mitchell, C.M.; Baugh, M.E.; Davy, K.P.; Neilson, A.P. A Mediterranean diet does not alter plasma trimethylamine N-oxide concentrations in healthy adults at risk for colon cancer. Food Funct. 2019, 10, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Casso, A.G.; VanDongen, N.S.; Ziemba, B.P.; Sapinsley, Z.J.; Richey, J.J.; Zigler, M.C.; Neilson, A.P.; Davy, K.P.; et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension 2020, 76, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Li, X.S.; Fan, Y.; Li, D.S.; Wu, Y.; Hazen, S.L. Increased Trimethylamine N-Oxide Portends High Mortality Risk Independent of Glycemic Control in Patients with Type 2 Diabetes Mellitus. Clin. Chem. 2017, 63, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafune, A.; Iwamoto, T.; Tsutsumi, Y.; Nakashima, A.; Yamamoto, I.; Yokoyama, K.; Yokoo, T.; Urashima, M. Associations among serum trimethylamine-N-oxide (TMAO) levels, kidney function and infarcted coronary artery number in patients undergoing cardiovascular surgery: A cross-sectional study. Clin. Exp. Nephrol. 2016, 20, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Missailidis, C.; Hallqvist, J.; Qureshi, A.R.; Barany, P.; Heimburger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, G.A.; Johansen, K.L.; Chertow, G.M.; Dalrymple, L.S.; Kornak, J.; Grimes, B.; Dwyer, T.; Chassy, A.W.; Fiehn, O. Associations of Trimethylamine N-Oxide with Nutritional and Inflammatory Biomarkers and Cardiovascular Outcomes in Patients New to Dialysis. J. Ren. Nutr. 2015, 25, 351–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutagy, N.E.; Neilson, A.P.; Osterberg, K.L.; Smithson, A.T.; Englund, T.R.; Davy, B.M.; Hulver, M.W.; Davy, K.P. Short-term high-fat diet increases postprandial trimethylamine-N-oxide in humans. Nutr. Res. 2015, 35, 858–864. [Google Scholar] [CrossRef]
- Wang, Z.; Levison, B.S.; Hazen, J.E.; Donahue, L.; Li, X.M.; Hazen, S.L. Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal. Biochem. 2014, 455, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Ilozumba, M.N.; Cheng, T.D.; Neuhouser, M.L.; Miller, J.W.; Beresford, S.A.A.; Duggan, D.J.; Toriola, A.T.; Song, X.; Zheng, Y.; Bailey, L.B.; et al. Associations between Plasma Choline Metabolites and Genetic Polymorphisms in One-Carbon Metabolism in Postmenopausal Women: The Women’s Health Initiative Observational Study. J. Nutr. 2020, 150, 2874–2881. [Google Scholar] [CrossRef]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Xu, M.; McCanna, D.J.; Sivak, J.G. Use of the viability reagent PrestoBlue in comparison with alamarBlue and MTT to assess the viability of human corneal epithelial cells. J. Pharmacol. Toxicol. Methods 2015, 71, 1–7. [Google Scholar] [CrossRef]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [CrossRef] [PubMed]
- Draney, C.; Hobson, A.E.; Grover, S.G.; Jack, B.O.; Tessem, J.S. Cdk5r1 Overexpression Induces Primary β-Cell Proliferation. J. Diabetes Res. 2016, 2016, 6375804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauchle, C.J.; Rohli, K.E.; Boyer, C.K.; Pal, V.; Rocheleau, J.V.; Liu, S.; Imai, Y.; Taylor, E.B.; Stephens, S.B. Mitochondrial Efflux of Citrate and Isocitrate Is Fully Dispensable for Glucose-Stimulated Insulin Secretion and Pancreatic Islet β-Cell Function. Diabetes 2021, 70, 1717–1728. [Google Scholar] [CrossRef]
- Truchan, N.A.; Brar, H.K.; Gallagher, S.J.; Neuman, J.C.; Kimple, M.E. A single-islet microplate assay to measure mouse and human islet insulin secretion. Islets 2015, 7, e1076607. [Google Scholar] [CrossRef] [Green Version]
- Zhao, A.; Ohara-Imaizumi, M.; Brissova, M.; Benninger, R.K.; Xu, Y.; Hao, Y.; Abramowitz, J.; Boulay, G.; Powers, A.C.; Piston, D.; et al. Gαo represses insulin secretion by reducing vesicular docking in pancreatic β-cells. Diabetes 2010, 59, 2522–2529. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gong, Y.; Li, C.; Zu, Y.; Cui, S.; Wan, L.; Chen, X. Pericentrin expression in pancreatic β cells is associated impaired glucose tolerance. Am. J. Transl. Res. 2019, 11, 2257–2268. [Google Scholar] [PubMed]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [CrossRef]
- Rowley, T.J., IV; Bitner, B.F.; Ray, J.D.; Lathen, D.R.; Smithson, A.T.; Dallon, B.W.; Plowman, C.J.; Bikman, B.T.; Hansen, J.M.; Dorenkott, M.R.; et al. Monomeric cocoa catechins enhance β-cell function by increasing mitochondrial respiration. J. Nutr. Biochem. 2017, 49, 30–41. [Google Scholar] [CrossRef]
- Kirlin, W.G.; Cai, J.; Thompson, S.A.; Diaz, D.; Kavanagh, T.J.; Jones, D.P. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic. Biol. Med. 1999, 27, 1208–1218. [Google Scholar] [CrossRef]
- Harris, C.; Hansen, J.M. Oxidative stress, thiols, and redox profiles. Methods Mol. Biol. 2012, 889, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Draney, C.; Austin, M.C.; Leifer, A.H.; Smith, C.J.; Kener, K.B.; Aitken, T.J.; Hess, K.H.; Haines, A.C.; Lett, L.A.; Hernandez-Carretero, A.; et al. HDAC1 overexpression enhances β-cell proliferation by down-regulating Cdkn1b/p27. Biochem. J. 2018, 475, 3997–4010. [Google Scholar] [CrossRef]
- Cherrington, N.J.; Cao, Y.; Cherrington, J.W.; Rose, R.L.; Hodgson, E. Physiological factors affecting protein expression of flavin-containing monooxygenases 1, 3 and 5. Xenobiotica 1998, 28, 673–682. [Google Scholar] [CrossRef]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattard, V.; Lachuer, J.; Buronfosse, T.; Garnier, F.; Benoit, E. Physiological factors affecting the expression of FMO1 and FMO3 in the rat liver and kidney. Biochem. Pharmacol. 2002, 63, 1453–1464. [Google Scholar] [CrossRef]
- Jezek, P.; Jaburek, M.; Plecita-Hlavata, L. Contribution of Oxidative Stress and Impaired Biogenesis of Pancreatic β-Cells to Type 2 Diabetes. Antioxid. Redox Signal. 2019, 31, 722–751. [Google Scholar] [CrossRef] [Green Version]
- Achard, C.S.; Laybutt, D.R. Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: Adaptation with enhanced insulin signaling or insulin resistance. Endocrinology 2012, 153, 2164–2177. [Google Scholar] [CrossRef]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladriere, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Erratum to: Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2012, 55, 863. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.; Chen, H.; Zhang, H.; Wan, X.; Su, Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine 2012, 42, 107–117. [Google Scholar] [CrossRef]
- Lin, N.; Zhang, H.; Su, Q. Advanced glycation end-products induce injury to pancreatic beta cells through oxidative stress. Diabetes Metab. 2012, 38, 250–257. [Google Scholar] [CrossRef]
- Rocha, M.; Diaz-Morales, N.; Rovira-Llopis, S.; Escribano-Lopez, I.; Banuls, C.; Hernandez-Mijares, A.; Diamanti-Kandarakis, E.; Victor, V.M. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Diabetes. Curr. Pharm. Des. 2016, 22, 2640–2649. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Sigfrid, L.A.; Cunningham, J.M.; Beeharry, N.; Hakan Borg, L.A.; Rosales Hernandez, A.L.; Carlsson, C.; Bone, A.J.; Green, I.C. Antioxidant enzyme activity and mRNA expression in the islets of Langerhans from the BB/S rat model of type 1 diabetes and an insulin-producing cell line. J. Mol. Med. 2004, 82, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Grankvist, K.; Marklund, S.L.; Taljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef]
- Unger, R.H.; Grundy, S. Hyperglycaemia as an inducer as well as a consequence of impaired islet cell function and insulin resistance: Implications for the management of diabetes. Diabetologia 1985, 28, 119–121. [Google Scholar] [CrossRef]
- Kleinfeld, A.M.; Prothro, D.; Brown, D.L.; Davis, R.C.; Richieri, G.V.; DeMaria, A. Increases in serum unbound free fatty acid levels following coronary angioplasty. Am. J. Cardiol. 1996, 78, 1350–1354. [Google Scholar] [CrossRef]
- Akerfeldt, M.C.; Howes, J.; Chan, J.Y.; Stevens, V.A.; Boubenna, N.; McGuire, H.M.; King, C.; Biden, T.J.; Laybutt, D.R. Cytokine-induced β-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 2008, 57, 3034–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela-Guajardo, J.E.; Garza-Gonzalez, S.; Garcia, N. Glucolipotoxicity-induced Oxidative Stress is Related to Mitochondrial Dysfunction and Apoptosis of Pancreatic β-cell. Curr. Diabetes Rev. 2021, 17, e031120187541. [Google Scholar] [CrossRef]
- Wang, H.; Kouri, G.; Wollheim, C.B. ER stress and SREBP-1 activation are implicated in β-cell glucolipotoxicity. J. Cell Sci. 2005, 118, 3905–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. A switch from life to death in endoplasmic reticulum stressed β-cells. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 58–65. [Google Scholar] [CrossRef] [Green Version]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with β-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef] [Green Version]
- Somesh, B.P.; Verma, M.K.; Sadasivuni, M.K.; Mammen-Oommen, A.; Biswas, S.; Shilpa, P.C.; Reddy, A.K.; Yateesh, A.N.; Pallavi, P.M.; Nethra, S.; et al. Chronic glucolipotoxic conditions in pancreatic islets impair insulin secretion due to dysregulated calcium dynamics, glucose responsiveness and mitochondrial activity. BMC Cell Biol. 2013, 14, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, B.J.; Mastronarde, D.N.; Buttle, K.F.; Howell, K.E.; McIntosh, J.R. Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc. Natl. Acad. Sci. USA 2001, 98, 2399–2406. [Google Scholar] [CrossRef] [Green Version]
- Steiner, D.F. The proprotein convertases. Curr. Opin. Chem. Biol. 1998, 2, 31–39. [Google Scholar] [CrossRef]
- Davidson, H.W. (Pro)Insulin processing: A historical perspective. Cell Biochem. Biophys. 2004, 40, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Davidson, H.W.; Rhodes, C.J.; Hutton, J.C. Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic β cell via two distinct site-specific endopeptidases. Nature 1988, 333, 93–96. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Zhang, J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell. Signal. 2016, 28, 1099–1104. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Haataja, L.; Gurlo, T.; Butler, A.E.; Wu, X.; Soeller, W.C.; Butler, P.C. Induction of endoplasmic reticulum stress-induced β-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1656–E1662. [Google Scholar] [CrossRef]
- Pirot, P.; Naamane, N.; Libert, F.; Magnusson, N.E.; Orntoft, T.F.; Cardozo, A.K.; Eizirik, D.L. Global profiling of genes modified by endoplasmic reticulum stress in pancreatic beta cells reveals the early degradation of insulin mRNAs. Diabetologia 2007, 50, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusta, B.; Baggio, L.L.; Estall, J.L.; Koehler, J.A.; Holland, D.P.; Li, H.; Pipeleers, D.; Ling, Z.; Drucker, D.J. GLP-1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006, 4, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Cnop, M. ER stress in pancreatic β cells: The thin red line between adaptation and failure. Sci. Signal. 2010, 3, pe7. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Mao, T.; Zhang, Y.; Shao, M.; You, J.; Ding, Q.; Chen, Y.; Wu, D.; Xie, D.; Lin, X.; et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1α activation in pancreatic β cells. Sci. Signal. 2010, 3, ra7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Saito, M.; Kadokura, H.; Miyazaki, J.I.; Tashiro, F.; Imagawa, Y.; Iwawaki, T.; Kohno, K. IRE1-XBP1 pathway regulates oxidative proinsulin folding in pancreatic β cells. J. Cell Biol. 2018, 217, 1287–1301. [Google Scholar] [CrossRef]
- Parkin, K.L.; Hultin, H.O. Partial purification of trimethylamine-N-oxide (TMAO) demethylase from crude fish muscle microsomes by detergents. J. Biochem. 1986, 100, 87–97. [Google Scholar] [CrossRef]
- Ganguly, P.; Boserman, P.; van der Vegt, N.F.A.; Shea, J.E. Trimethylamine N-oxide Counteracts Urea Denaturation by Inhibiting Protein-Urea Preferential Interaction. J. Am. Chem. Soc. 2018, 140, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.L.; Hyeon, C.; Thirumalai, D. Effects of trimethylamine N-oxide (TMAO) and crowding agents on the stability of RNA hairpins. J. Am. Chem. Soc. 2008, 130, 7364–7372. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Timasheff, S.N. The stabilization of proteins by osmolytes. Biophys. J. 1985, 47, 411–414. [Google Scholar] [CrossRef]
- Mashino, T.; Fridovich, I. Effects of urea and trimethylamine-N-oxide on enzyme activity and stability. Arch. Biochem. Biophys. 1987, 258, 356–360. [Google Scholar] [CrossRef]
- Hu, C.Y.; Lynch, G.C.; Kokubo, H.; Pettitt, B.M. Trimethylamine N-oxide influence on the backbone of proteins: An oligoglycine model. Proteins 2010, 78, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Bennion, B.J.; DeMarco, M.L.; Daggett, V. Preventing misfolding of the prion protein by trimethylamine N-oxide. Biochemistry 2004, 43, 12955–12963. [Google Scholar] [CrossRef] [PubMed]
- Bennion, B.J.; Daggett, V. Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: A chemical chaperone at atomic resolution. Proc. Natl. Acad. Sci. USA 2004, 101, 6433–6438. [Google Scholar] [CrossRef] [Green Version]
- Mulhern, M.L.; Madson, C.J.; Kador, P.F.; Randazzo, J.; Shinohara, T. Cellular osmolytes reduce lens epithelial cell death and alleviate cataract formation in galactosemic rats. Mol. Vis. 2007, 13, 1397–1405. [Google Scholar] [PubMed]
- Rezus, Y.L.; Bakker, H.J. Destabilization of the hydrogen-bond structure of water by the osmolyte trimethylamine N-oxide. J. Phys. Chem. B 2009, 113, 4038–4044. [Google Scholar] [CrossRef]
- Yancey, P.H.; Siebenaller, J.F. Co-evolution of proteins and solutions: Protein adaptation versus cytoprotective micromolecules and their roles in marine organisms. J. Exp. Biol. 2015, 218, 1880–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Xiong, S. TMAO-Protein Preferential Interaction Profile Determines TMAO’s Conditional In Vivo Compatibility. Biophys. J. 2016, 111, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Govindarajulu, M.; Pinky, P.D.; Steinke, I.; Bloemer, J.; Ramesh, S.; Kariharan, T.; Rella, R.T.; Bhattacharya, S.; Dhanasekaran, M.; Suppiramaniam, V.; et al. Gut Metabolite TMAO Induces Synaptic Plasticity Deficits by Promoting Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2020, 13, 138. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, X.; Xu, J.; Xue, C.; Xue, Y.; Wang, Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 2014, 118, 476–481. [Google Scholar] [CrossRef]
- Gao, X.; Xu, J.; Jiang, C.; Zhang, Y.; Xue, Y.; Li, Z.; Wang, J.; Xue, C.; Wang, Y. Fish oil ameliorates trimethylamine N-oxide-exacerbated glucose intolerance in high-fat diet-fed mice. Food Funct. 2015, 6, 1117–1125. [Google Scholar] [CrossRef]
- Mari, A. Mathematical modeling in glucose metabolism and insulin secretion. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, T.J.; Lee, J.C.; Choi, K.; Kim, M.Y.; Kim, H.C.; Cho, Y.M.; Kim, S. Simulation of oral glucose tolerance tests and the corresponding isoglycemic intravenous glucose infusion studies for calculation of the incretin effect. J. Korean Med. Sci. 2014, 29, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalowska, J.; Miller-Kasprzak, E.; Bogdanski, P. Incretin Hormones in Obesity and Related Cardiometabolic Disorders: The Clinical Perspective. Nutrients 2021, 13, 351. [Google Scholar] [CrossRef] [PubMed]
- Creutzfeldt, W. The incretin concept today. Diabetologia 1979, 16, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Creutzfeldt, W.; Ebert, R. New developments in the incretin concept. Diabetologia 1985, 28, 565–573. [Google Scholar] [CrossRef]
- Meier, J.J.; Nauck, M.A. Glucagon-like peptide 1(GLP-1) in biology and pathology. Diabetes Metab. Res. Rev. 2005, 21, 91–117. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Bergman, R.N.; Phillips, L.S.; Cobelli, C. Physiologic evaluation of factors controlling glucose tolerance in man: Measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J. Clin. Investig. 1981, 68, 1456–1467. [Google Scholar] [CrossRef] [Green Version]
- Dumas, M.E.; Rothwell, A.R.; Hoyles, L.; Aranias, T.; Chilloux, J.; Calderari, S.; Noll, E.M.; Pean, N.; Boulange, C.L.; Blancher, C.; et al. Microbial-Host Co-metabolites Are Prodromal Markers Predicting Phenotypic Heterogeneity in Behavior, Obesity, and Impaired Glucose Tolerance. Cell Rep. 2017, 20, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.M.; McManus, S.A.; Hughes, W.E.; Schmitz-Peiffer, C. Flavin-Containing Monooxygenase 3 Reduces Endoplasmic Reticulum Stress in Lipid-Treated Hepatocytes. Mol. Endocrinol. 2016, 30, 417–428. [Google Scholar] [CrossRef]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Costa, S.; Sorenson, M.M.; Sola-Penna, M. Counteracting effects of urea and methylamines in function and structure of skeletal muscle myosin. Arch. Biochem. Biophys. 2002, 408, 272–278. [Google Scholar] [CrossRef]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupachyk, S.; Watcho, P.; Stavniichuk, R.; Shevalye, H.; Obrosova, I.G. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes 2013, 62, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, R.; Ge, X.; Han, L.; Yu, P.; Gong, X.; Meng, Q.; Zhang, Y.; Fan, H.; Zheng, L.; Liu, Z.; et al. Gut microbe-generated metabolite trimethylamine N-oxide and the risk of diabetes: A systematic review and dose-response meta-analysis. Obes. Rev. 2019, 20, 883–894. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Q.; Jiang, H. Gut microbiota in atherosclerosis: Focus on trimethylamine N-oxide. APMIS 2020, 128, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Wang, Z.; Tang, W.H.W.; Hazen, S.L. Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline Is Prothrombotic in Subjects. Circulation 2017, 135, 1671–1673. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, Z. Impact of trimethylamine N-oxide (TMAO) metaorganismal pathway on cardiovascular disease. J. Lab. Precis. Med. 2020, 5, 16. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, C.I.; Vallejo, J.A.; Wang, D.; Gray, M.A.; Tiede-Lewis, L.M.; Shawgo, T.; Daon, E.; Zorn, G., 3rd; Stubbs, J.R.; Wacker, M.J. Trimethylamine-N-oxide acutely increases cardiac muscle contractility. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1272–H1282. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Nowinski, A. Is increased plasma TMAO a compensatory response to hydrostatic and osmotic stress in cardiovascular diseases? Med. Hypotheses 2019, 130, 109271. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krueger, E.S.; Beales, J.L.; Russon, K.B.; Elison, W.S.; Davis, J.R.; Hansen, J.M.; Neilson, A.P.; Hansen, J.M.; Tessem, J.S. Gut Metabolite Trimethylamine N-Oxide Protects INS-1 β-Cell and Rat Islet Function under Diabetic Glucolipotoxic Conditions. Biomolecules 2021, 11, 1892. https://doi.org/10.3390/biom11121892
Krueger ES, Beales JL, Russon KB, Elison WS, Davis JR, Hansen JM, Neilson AP, Hansen JM, Tessem JS. Gut Metabolite Trimethylamine N-Oxide Protects INS-1 β-Cell and Rat Islet Function under Diabetic Glucolipotoxic Conditions. Biomolecules. 2021; 11(12):1892. https://doi.org/10.3390/biom11121892
Chicago/Turabian StyleKrueger, Emily S., Joseph L. Beales, Kacie B. Russon, Weston S. Elison, Jordan R. Davis, Jackson M. Hansen, Andrew P. Neilson, Jason M. Hansen, and Jeffery S. Tessem. 2021. "Gut Metabolite Trimethylamine N-Oxide Protects INS-1 β-Cell and Rat Islet Function under Diabetic Glucolipotoxic Conditions" Biomolecules 11, no. 12: 1892. https://doi.org/10.3390/biom11121892
APA StyleKrueger, E. S., Beales, J. L., Russon, K. B., Elison, W. S., Davis, J. R., Hansen, J. M., Neilson, A. P., Hansen, J. M., & Tessem, J. S. (2021). Gut Metabolite Trimethylamine N-Oxide Protects INS-1 β-Cell and Rat Islet Function under Diabetic Glucolipotoxic Conditions. Biomolecules, 11(12), 1892. https://doi.org/10.3390/biom11121892