
Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis

Abstract
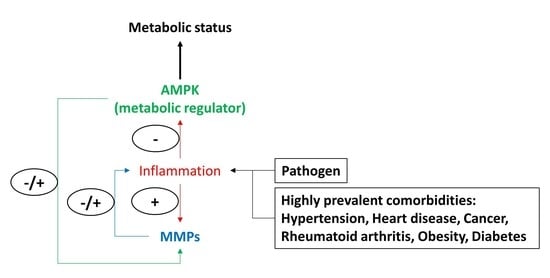
:
1. Introduction
2. MMPs and Their Implications in Pulmonary Pathologies
3. COVID-19 Clinical Characteristics
3.1. COVID-19 Severity Spectrum May Be Related to Metabolic Changes in the Lung and Extra-Pulmonary Organs
3.2. Metabolic Alterations in the Lungs
3.3. Metabolic Alterations in Extra-Pulmonary Organs
4. MMP-Involvement in Organ Damage Warrants Investigation
Role of MMPs, Inflammatory Response, and AMPK
5. Improving Metabolism Through Manipulation of the Possibly Synergic or Cooperative Actions of MMPs, Cytokines, and AMPK Pathways on Systemic Metabolism
6. Limitations
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Contribution to the Field Statement
References
- Ayres, J.S. Surviving COVID-19: A disease tolerance perspective. Sci. Adv. 2020, 6, eabc1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, D.S.; Ayres, J.S. Two ways to survive infection: What resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 2008, 8, 889–895. [Google Scholar] [CrossRef]
- Ayres, J.S. A metabolic handbook for the COVID-19 pandemic. Nat. Metab. 2020, 2, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, R.; Carlos, A.R.; Braza, F.; Thompson, J.A.; Bastos-Amador, P.; Ramos, S.; Soares, M.P. Disease Tolerance as an Inherent Component of Immunity. Annu. Rev. Immunol. 2019, 37, 405–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumoto, K.; Kagaya, K.; Zarebski, A.; Chowell, G. Estimating the asymptomatic proportion of coronavirus disease 2019 (COVID-19) cases on board the Diamond Princess cruise ship, Yokohama, Japan, 2020. Eurosurveillance 2020, 25, 2000180. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, Q.; Jia, C.; Wang, W.; Chen, J.; Xia, Y.; Wang, X.; Wen, M.; Wang, H.; Zhang, Z.; et al. Epidemiological Characteristics and Clinical Outcomes of Coronavirus Disease Patients in Northwest China: High-Volume Research from Low Population Density Regions. Front. Med. 2020, 7, 564250. [Google Scholar] [CrossRef]
- Liu, L.; Lei, X.; Xiao, X.; Yang, J.; Li, J.; Ji, M.; Du, W.; Tan, H.; Zhu, J.; Li, B.; et al. Epidemiological and Clinical Characteristics of Patients with Coronavirus Disease-2019 in Shiyan City, China. Front. Cell. Infect. Microbiol. 2020, 10, 284. [Google Scholar] [CrossRef]
- Lei, Z.; Cao, H.; Jie, Y.; Huang, Z.; Guo, X.; Chen, J.; Peng, L.; Dai, X.; Liu, J.; Li, X.; et al. A cross-sectional comparison of epidemiological and clinical features of patients with coronavirus disease (COVID-19) in Wuhan and outside Wuhan, China. Travel Med. Infect. Dis. 2020, 35, 101664. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Xie, J.; Wu, W.; Li, S.; Hu, Y.; Hu, M.; Li, J.; Yang, Y.; Huang, T.; Zheng, K.; Wang, Y.; et al. Clinical characteristics and outcomes of critically ill patients with novel coronavirus infectious disease (COVID-19) in China: A retrospective multicenter study. Intensiv. Care Med. 2020, 46, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal. Transduct. Target. Ther. 2020, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Patron, C.; Kassiri, Z.; Leung, D. Modulation of Systemic Metabolism by MMP-2: From MMP-2 Deficiency in Mice to MMP-2 Deficiency in Patients. Compr. Physiol. 2016, 6, 1935–1949. [Google Scholar] [PubMed]
- Oikonomidi, S.; Kostikas, K.; Tsilioni, I.; Tanou, K.; Gourgoulianis, K.I.; Kiropoulos, T.S. Matrix metalloproteinases in respiratory diseases: From pathogenesis to potential clinical implications. Curr. Med. Chem. 2009, 16, 1214–1228. [Google Scholar] [CrossRef]
- Elkington, P.T.; Friedland, J.S. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006, 61, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Dejonckheere, E.; Libert, C. A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur. Respir. J. 2011, 38, 1200–1214. [Google Scholar] [CrossRef]
- Davey, A.; McAuley, D.F.; O’Kane, C.M. Matrix metalloproteinases in acute lung injury: Mediators of injury and drivers of repair. Eur. Respir. J. 2011, 38, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, D.K.; Shido, K.; Kopp, H.G.; Petit, I.; Shmelkov, S.V.; Young, L.M.; Hooper, A.T.; Amano, H.; Avecilla, S.T.; Heissig, B.; et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat. Med. 2006, 12, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.; Yi, J.; Guo, A.; Wang, X.; Overall, C.M.; Jiang, W.; Elde, R.; Borregaard, N.; Pei, D. Subcellular distribution and cytokine- and chemokine-regulated secretion of leukolysin/MT6-MMP/MMP-25 in neutrophils. J. Biol. Chem. 2001, 276, 21960–21968. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Morrison, C.J.; Butler, G.S.; Rodriguez, D.; Overall, C.M. Matrix metalloproteinase proteomics: Substrates, targets, and therapy. Curr. Opin. Cell Biol. 2009, 21, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, F.; Corry, D.B. Discovery of novel markers in allergic lung inflammation through proteomic-based technologies. Expert Rev. Proteom. 2008, 5, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenlee, K.J.; Werb, Z.; Kheradmand, F. Matrix metalloproteinases in lung: Multiple, multifarious, and multifaceted. Physiol. Rev. 2007, 87, 69–98. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, K.J.; Corry, D.B.; Engler, D.A.; Matsunami, R.K.; Tessier, P.; Cook, R.G.; Werb, Z.; Kheradmand, F. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J. Immunol. 2006, 177, 7312–7321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corry, D.B.; Rishi, K.; Kanellis, J.; Kiss, A.; Song, L.Z.; Xu, J.; Feng, L.; Werb, Z.; Kheradmand, F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat. Immunol. 2002, 3, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Corry, D.B.; Kiss, A.; Song, L.Z.; Song, L.; Xu, J.; Lee, S.H.; Werb, Z.; Kheradmand, F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004, 18, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Holter, J.C.; Holten, A.R.; Muller, K.E.; Lind, A.; Bekken, G.K.; Dudman, S.; Aukrust, P.; Dyrhol-Riise, A.M.; Heggelund, L. Distinct and early increase in circulating MMP-9 in COVID-19 patients with respiratory failure. J. Infect. 2020, 81, e41–e43. [Google Scholar] [CrossRef]
- Ohbayashi, H. Matrix metalloproteinases in lung diseases. Curr. Protein Pept. Sci. 2002, 3, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Gueders, M.M.; Foidart, J.M.; Noel, A.; Cataldo, D.D. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: Potential implications in asthma and other lung diseases. Eur. J. Pharm. 2006, 533, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Lagente, V.; Manoury, B.; Nenan, S.; Le Quement, C.; Martin-Chouly, C.; Boichot, E. Role of matrix metalloproteinases in the development of airway inflammation and remodeling. Braz. J. Med. Biol. Res. 2005, 38, 1521–1530. [Google Scholar] [CrossRef] [Green Version]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (Bethesda) 2013, 28, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demedts, I.K.; Brusselle, G.G.; Bracke, K.R.; Vermaelen, K.Y.; Pauwels, R.A. Matrix metalloproteinases in asthma and COPD. Curr. Opin. Pharm. 2005, 5, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Ricou, B.; Nicod, L.; Lacraz, S.; Welgus, H.G.; Suter, P.M.; Dayer, J.M. Matrix metalloproteinases and TIMP in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996, 154 Pt 1, 346–352. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proc. Am. Thorac. Soc. 2006, 3, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Zhang, X.; Drelich, A.; Shi, J.; Hsu, J.C.; Luchsinger, L.; Hillyer, C.D.; Tseng, C.K.; Jiang, S.; Du, L. A novel receptor-binding domain (RBD)-based mRNA vaccine against SARS-CoV-2. Cell Res. 2020, 30, 932–935. [Google Scholar] [CrossRef]
- Yang, J.; Wang, W.; Chen, Z.; Lu, S.; Yang, F.; Bi, Z.; Bao, L.; Mo, F.; Li, X.; Huang, Y.; et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 2020, 586, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 1996, 334, 1469–1470. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The clinical pathology of severe acute respiratory syndrome (SARS): A report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J. Infect. 2020, 81, e6–e12. [Google Scholar] [CrossRef]
- Shi, H.; Han, X.; Jiang, N.; Cao, Y.; Alwalid, O.; Gu, J.; Fan, Y.; Zheng, C. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: A descriptive study. Lancet Infect. Dis. 2020, 20, 425–434. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase—Fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Evans, A.M.; Hardie, D.G.; Galione, A.; Peers, C.; Kumar, P.; Wyatt, C.N. AMP-activated protein kinase couples mitochondrial inhibition by hypoxia to cell-specific Ca2+ signalling mechanisms in oxygen-sensing cells. In Signalling Pathways in Acute Oxygen Sensing: Novartis Foundation Symposium; Wiley: Hoboken, NJ, USA, 2006; Volume 272, pp. 234–252. [Google Scholar]
- Holmes, B.F.; Sparling, D.P.; Olson, A.L.; Winder, W.W.; Dohm, G.L. Regulation of muscle GLUT4 enhancer factor and myocyte enhancer factor 2 by AMP-activated protein kinase. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E1071–E1076. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Soltys, C.L.; Young, M.E.; Proud, C.G.; Dyck, J.R. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J. Biol. Chem. 2004, 279, 32771–32779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyck, J.R.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574 Pt 1, 95–112. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Mailleux, F.; Gelinas, R.; Beauloye, C.; Horman, S.; Bertrand, L. O-GlcNAcylation, enemy or ally during cardiac hypertrophy development? Biochim. Biophys. Acta 2016, 1862, 2232–2243. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Wang, Q.; Coughlan, K.; Viollet, B.; Moriasi, C.; Zou, M.H. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J. Pathol. 2013, 182, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Otsuki, T.; Kozu, K.; Numano, K.; Suzuki, K.; et al. Protective Roles of Endothelial AMP-Activated Protein Kinase Against Hypoxia-Induced Pulmonary Hypertension in Mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zmijewski, J.W.; Lorne, E.; Liu, G.; Park, Y.J.; Tsuruta, Y.; Abraham, E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L497–L504. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, L.; Locatelli, F. Hypoxia response and acute lung and kidney injury: Possible implications for therapy of COVID-19. Clin. Kidney J. 2020, 13, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Reis, T. Kidney involvement in COVID-19 and rationale for extracorporeal therapies. Nat. Rev. Nephrol. 2020, 16, 308–310. [Google Scholar] [CrossRef] [Green Version]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Ardehali, H.; Sabbah, H.N.; Burke, M.A.; Sarma, S.; Liu, P.P.; Cleland, J.G.; Maggioni, A.; Fonarow, G.C.; Abel, E.D.; Campia, U.; et al. Targeting myocardial substrate metabolism in heart failure: Potential for new therapies. Eur. J. Heart Fail. 2012, 14, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Chavez, P.N.; Stanley, W.C.; McElfresh, T.A.; Huang, H.; Sterk, J.P.; Chandler, M.P. Effect of hyperglycemia and fatty acid oxidation inhibition during aerobic conditions and demand-induced ischemia. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1521–H1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favalli, E.G.; Caporali, R. GM-CSF in the treatment of COVID-19: A new conductor in the pathogenesis of cytokine storm? Lancet Rheumatol. 2020, 2, e448–e449. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagunduz, P.; Tabak, F.; Atagunduz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, E.; Fernandez-Patron, C. Destroy to Rebuild: The Connection Between Bone Tissue Remodeling and Matrix Metalloproteinases. Front. Physiol. 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Hardy-Rando, E.; Fernandez-Patron, C. Emerging pathways of communication between the heart and non-cardiac organs. J. Biomed. Res. 2019, 33, 145–155. [Google Scholar]
- Hardy, E.; Hardy-Sosa, A.; Fernandez-Patron, C. MMP-2: Is too low as bad as too high in the cardiovascular system? Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1332–H1340. [Google Scholar] [CrossRef]
- Toba, H.; Cannon, P.L.; Yabluchanskiy, A.; Iyer, R.P.; D’Armiento, J.; Lindsey, M.L. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H375–H383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [CrossRef]
- Rajavashisth, T.B.; Liao, J.K.; Galis, Z.S.; Tripathi, S.; Laufs, U.; Tripathi, J.; Chai, N.N.; Xu, X.P.; Jovinge, S.; Shah, P.K.; et al. Inflammatory cytokines and oxidized low density lipoproteins increase endothelial cell expression of membrane type 1-matrix metalloproteinase. J. Biol. Chem. 1999, 274, 11924–11929. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Gorgun, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, M.C.L.; Herlea-Pana, O.; Brobst, D.; Yang, X.; Wood, J.; Hu, X.; Liu, Z.; Lee, C.W.; Zaw, A.M.; Chow, B.K.C.; et al. Tumor Necrosis Factor-alpha Promotes Phosphoinositide 3-Kinase Enhancer A and AMP-Activated Protein Kinase Interaction to Suppress Lipid Oxidation in Skeletal Muscle. Diabetes 2017, 66, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, L.K.; Jones, L.R.; Cohen, P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. 1991, 196, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Unger, R.H. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E216–E221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Nocon, A.; Fry, J.; Sherban, A.; Rui, X.; Jiang, B.; Xu, X.J.; Han, J.; Yan, Y.; Yang, Q.; et al. AMPK Activation by Metformin Suppresses Abnormal Extracellular Matrix Remodeling in Adipose Tissue and Ameliorates Insulin Resistance in Obesity. Diabetes 2016, 65, 2295–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizane, Y.; Thanos, A.; Takeuchi, K.; Murakami, Y.; Kayama, M.; Trichonas, G.; Miller, J.; Foretz, M.; Viollet, B.; Vavvas, D.G. AMP-activated protein kinase suppresses matrix metalloproteinase-9 expression in mouse embryonic fibroblasts. J. Biol. Chem. 2011, 286, 16030–16038. [Google Scholar] [CrossRef] [Green Version]
- Dadson, K.; Chasiotis, H.; Wannaiampikul, S.; Tungtrongchitr, R.; Xu, A.; Sweeney, G. Adiponectin mediated APPL1-AMPK signaling induces cell migration, MMP activation, and collagen remodeling in cardiac fibroblasts. J. Cell. Biochem. 2014, 115, 785–793. [Google Scholar] [CrossRef]
- Allenbach, Y.; Saadoun, D.; Maalouf, G.; Vieira, M.; Hellio, A.; Boddaert, J.; Gros, H.; Salem, J.E.; Resche Rigon, M.; Menyssa, C.; et al. Development of a multivariate prediction model of intensive care unit transfer or death: A French prospective cohort study of hospitalized COVID-19 patients. PLoS ONE 2020, 15, e0240711. [Google Scholar]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Abers, M.S.; Delmonte, O.M.; Ricotta, E.E.; Fintzi, J.; Fink, D.L.; de Jesus, A.A.A.; Zarember, K.A.; Alehashemi, S.; Oikonomou, V.; Desai, J.V.; et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight 2021, 6, e144455. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.D.; Yaffe, M.B. COVID-19: All the wrong moves in all the wrong places. Sci. Signal. 2020, 13, eabe4242. [Google Scholar] [CrossRef]
- Atkinson, J.J.; Senior, R.M. Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 2003, 28, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Timms, P.M.; Mannan, N.; Hitman, G.A.; Noonan, K.; Mills, P.G.; Syndercombe-Court, D.; Aganna, E.; Price, C.P.; Boucher, B.J. Circulating MMP9, vitamin D and variation in the TIMP-1 response with VDR genotype: Mechanisms for inflammatory damage in chronic disorders? QJM 2002, 95, 787–796. [Google Scholar] [CrossRef]
- Coussens, A.; Timms, P.M.; Boucher, B.J.; Venton, T.R.; Ashcroft, A.T.; Skolimowska, K.H.; Newton, S.M.; Wilkinson, K.A.; Davidson, R.N.; Griffiths, C.J.; et al. 1alpha,25-dihydroxyvitamin D3 inhibits matrix metalloproteinases induced by Mycobacterium tuberculosis infection. Immunology 2009, 127, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sakamuri, S.; Watts, R.; Takawale, A.; Wang, X.; Hernandez-Anzaldo, S.; Bahitham, W.; Fernandez-Patron, C.; Lehner, R.; Kassiri, Z. Absence of Tissue Inhibitor of Metalloproteinase-4 (TIMP4) ameliorates high fat diet-induced obesity in mice due to defective lipid absorption. Sci. Rep. 2017, 7, 6210. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Anzaldo, S.; Brglez, V.; Hemmeryckx, B.; Leung, D.; Filep, J.G.; Vance, J.E.; Vance, D.E.; Kassiri, Z.; Lijnen, R.H.; Lambeau, G.; et al. Novel Role for Matrix Metalloproteinase 9 in Modulation of Cholesterol Metabolism. J. Am. Heart Assoc. 2016, 5, e004228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Berry, E.; Hernandez-Anzaldo, S.; Takawale, A.; Kassiri, Z.; Fernandez-Patron, C. Matrix metalloproteinase-2 mediates a mechanism of metabolic cardioprotection consisting of negative regulation of the sterol regulatory element-binding protein-2/3-hydroxy-3-methylglutaryl-CoA reductase pathway in the heart. Hypertension 2015, 65, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Berry, E.; Hernandez-Anzaldo, S.; Sun, D.; Adijiang, A.; Li, L.; Zhang, D.; Fernandez-Patron, C. MMP-2 inhibits PCSK9-induced degradation of the LDL receptor in Hepa1-c1c7 cells. FEBS Lett. 2015, 589, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Anzaldo, S.; Berry, E.; Brglez, V.; Leung, D.; Yun, T.J.; Lee, J.S.; Filep, J.G.; Kassiri, Z.; Cheong, C.; Lambeau, G.; et al. Identification of a Novel Heart-Liver Axis: Matrix Metalloproteinase-2 Negatively Regulates Cardiac Secreted Phospholipase A2 to Modulate Lipid Metabolism and Inflammation in the Liver. J. Am. Heart Assoc. 2015, 4, e002553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, E.; Hernandez-Anzaldo, S.; Ghomashchi, F.; Lehner, R.; Murakami, M.; Gelb, M.H.; Kassiri, Z.; Wang, X.; Fernandez-Patron, C. Matrix metalloproteinase-2 negatively regulates cardiac secreted phospholipase A2 to modulate inflammation and fever. J. Am. Heart Assoc. 2015, 4, e001868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, M.P.; Armstrong, P.W.; Ruzyllo, W.; Brum, J.; Cusmano, L.; Krzeski, P.; Lyon, R.; Quinones, M.; Theroux, P.; Sydlowski, D.; et al. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: Results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J. Am. Coll. Cardiol. 2006, 48, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Cerisano, G.; Buonamici, P.; Gori, A.M.; Valenti, R.; Sciagra, R.; Giusti, B.; Sereni, A.; Raspanti, S.; Colonna, P.; Gensini, G.F.; et al. Matrix metalloproteinases and their tissue inhibitor after reperfused ST-elevation myocardial infarction treated with doxycycline. Insights from the TIPTOP trial. Int. J. Cardiol. 2015, 197, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.; Cao, J.; Chen, W.T. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 2000, 19, 6642–6650. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.E.A. Matrix Metalloproteinase Inhibition with Doxycycline to Prevent Adverse Remodeling After Acute Myocardial Infarction: A Pilot Study. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03508232 (accessed on 5 March 2021).
- Mohan, M.; Al-Talabany, S.; McKinnie, A.; Mordi, I.R.; Singh, J.S.S.; Gandy, S.J.; Baig, F.; Hussain, M.S.; Bhalraam, U.; Khan, F.; et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: The MET-REMODEL trial. Eur. Heart J. 2019, 40, 3409–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steneberg, P.; Lindahl, E.; Dahl, U.; Lidh, E.; Straseviciene, J.; Backlund, F.; Kjellkvist, E.; Berggren, E.; Lundberg, I.; Bergqvist, I.; et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 2018, 3, e99114. [Google Scholar] [CrossRef] [PubMed]
- Gomori, K.; Szabados, T.; Kenyeres, E.; Pipis, J.; Foldesi, I.; Siska, A.; Dorman, G.; Ferdinandy, P.; Gorbe, A.; Bencsik, P. Cardioprotective Effect of Novel Matrix Metalloproteinase Inhibitors. Int. J. Mol. Sci. 2020, 21, 6990. [Google Scholar] [CrossRef]
- Bosonea, A.M.; Wang, X.; Odenbach, J.; Fernandez-Patron, C. Metalloproteinases in hypertension and cardiac disease: Differential expression and mutual regulation. Drug Discov. Today Dis. Models 2011, 8, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chow, F.L.; Oka, T.; Hao, L.; Lopez-Campistrous, A.; Kelly, S.; Cooper, S.; Odenbach, J.; Finegan, B.A.; Schulz, R.; et al. Matrix metalloproteinase-7 and ADAM-12 (a disintegrin and metalloproteinase-12) define a signaling axis in agonist-induced hypertension and cardiac hypertrophy. Circulation 2009, 119, 2480–2489. [Google Scholar] [CrossRef] [Green Version]
- Odenbach, J.; Wang, X.; Cooper, S.; Chow, F.L.; Oka, T.; Lopaschuk, G.; Kassiri, Z.; Fernandez-Patron, C. MMP-2 mediates angiotensin II-induced hypertension under the transcriptional control of MMP-7 and TACE. Hypertension 2011, 57, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar]
- Iyer, R.P.; de Castro Bras, L.E.; Jin, Y.F.; Lindsey, M.L. Translating Koch’s postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: Cardiac metalloproteinase actions (CarMA) postulates. Circ. Res. 2014, 114, 860–871. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Desantis, S.M.; Baicu, C.F.; Stroud, R.E.; Thompson, S.B.; McClure, C.D.; Mehurg, S.M.; Spinale, F.G. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ. Heart Fail. 2011, 4, 246–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinale, F.G.; Villarreal, F. Targeting matrix metalloproteinases in heart disease: Lessons from endogenous inhibitors. Biochem. Pharm. 2014, 90, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.T.; Dyck, J.R. Is AMPK the savior of the failing heart? Trends Endocrinol. Metab. 2015, 26, 40–48. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. AMP-Activated Protein Kinase as a Reprogramming Strategy for Hypertension and Kidney Disease of Developmental Origin. Int. J. Mol. Sci. 2018, 19, 1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Lauhio, A.; Sorsa, T.; Lindy, O.; Suomalainen, K.; Saari, H.; Golub, L.M.; Konttinen, Y.T. The regulatory role of doxycycline/tetracycline in collagenolytic activity and tissue destruction in joint diseases: Comment on the article by Yu et al. Arthritis Rheum. 1993, 36, 1335–1336. [Google Scholar] [CrossRef]
- Golub, L.M.; Ciancio, S.; Ramamamurthy, N.S.; Leung, M.; McNamara, T.F. Low-dose doxycycline therapy: Effect on gingival and crevicular fluid collagenase activity in humans. J. Periodontal Res. 1990, 25, 321–330. [Google Scholar] [CrossRef]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Solun, B.; Shoenfeld, Y. Inhibition of metalloproteinases in therapy for severe lung injury due to COVID-19. Med. Drug Discov. 2020, 7, 100052. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| MMPs | Disease | Suggested MMP Role in Disease |
|---|---|---|
| MMP-1 | Emphysema, which is a chronic obstructive pulmonary disease that is characterized by extensive tissue remodeling, the destruction of small bronchi and alveolar septa, diminished lung plasticity, and ultimately impaired pulmonary oxygenation. Increased MMP-1 levels have been detected in type II pneumocyte in the lung parenchyma of patients with emphysema, as well as in alveolar lining cells and fibroblasts exposed to smoke. Asthma, which is a condition associated with lung inflammation, episodic dyspnea, and airway hyperresponsiveness. Increased MMP-1 levels have been detected in airway smooth muscle cells from asthma and emphysema patients. | Drives pulmonary tissue destruction such as the spontaneous development of air space expansion and emphysema-like changes including the coalescence of the alveolar spaces and disruption of the alveolar walls, as shown in transgenic animal models. Induces airway obstruction and ASMC hyperplasia in asthma by activating the IGF axis. Contributes to altered contractile response of smooth muscle cells induced by Th2 cytokines (IL-4 and IL-13), chiefly in allergic asthma. |
| MMP-2 | Asthma; increased MMP-2 levels have been detected in sputum of patients with this condition. Idiopathic pulmonary fibrosis (IPF), which is a chronic condition (usually lethal due to respiratory failure), characterized by disproportionate extracellular matrix degradation, inflammation, and fibrosis of the lung parenchyma. Pneumonia; increased MMP-2 levels have been detected in an elevated proportion of patients with this condition. | Participates in tissue degradation in emphysema. Aggravates pulmonary hypertension, which may lead to right heart failure and death. Involved in collagen deposition in IPF. |
| MMP-3 | Asthma; increased MMP-3 levels have been detected in bronchial lavages of individuals with this condition. | Involved in airway hyperreactivity and diminished lung function in asthmatic patients, which may be related to the activation of MMP-9 and the augmented synthesis of procollagen I. |
| MMP-7 | Asthma; increased MMP-7 levels have been detected in airway epithelial cells and in lung epithelial cells under the effect of osteopontin. Cystic fibrosis (CF), which is a genetic disorder characterized by malfunctioning of the chloride channels of the airway epithelia and other epithelial linings. Increased MMP-7 levels have been detected in type II pneumocytes in patients with CF | Contributes to airway epithelial damage and inflammation in subjects with severe asthma by cleaving and releasing soluble FasL. Plays a profibrotic effect in IPF. |
| MMP-8 | Exacerbated chronic obstructive pulmonary disease (COPD); COPD is a progressive pathology that has cigarette smoking as the main triggering factor, and is characterized by airway abnormal inflammation, airway flow obstruction, and pulmonary failure. Emphysema; increased MMP-8 levels have been detected in the bronchoalveolar lavage (BAL) fluid of smokers with this condition. Asthma; increased MMP-8 levels have been detected in the BAL fluid of patients with this condition. Pediatric severe chronic lung disease; increased MMP-8 levels have been detected in BAL fluids from preterm babies (before 28 weeks of gestation) who later developed this disease. | Increases collagenolytic activity, impairing the collagen network of the lung and thus contributing to emphysema in adults. Associated with severe granulocytic inflammation in airways caused by allergen exposure in MMP-8-deficient mice. Contributes to the lung harm that happens in the initial phases of chronic lung disease. Linked to bronchiectasis. Involved in severe chronic lung illness in preterm babies where the alveolarization of the saccules alveolaris is starting. Involved in pulmonary fibrosis. Triggers an inflammatory response in the lungs associated with injurious ventilation. |
| MMP-9 | COPD; increased MMP-9 levels have been detected in cells (e.g., alveolar macrophages) and sputum of patients with COPD (e.g., in bronchial epithelium and submucosal areas). Emphysema; increased MMP-9 levels have been detected in (i) the lung parenchyma of patients with this condition, (ii) alveolar macrophages from smokers, and (iii) cultured airway macrophages from smokers (at baseline and in response to IL-1b and LPS). Asthma; increased MMP-9 levels have been detected in (i) the sputum of subjects with allergic asthma after flour inhalation; (ii) the BAL fluid (mainly associated with airway neutrophils and less with alveolar macrophages—and maybe with eosinophils), sputum, serum, and bronchial tissues of asthmatic patients who have chronic submucosal inflammation; and (iii) samples from workers exposed to toluene diisocyanate-occupational asthma. Rapidly progressive IPF and IPF-usual interstitial pneumonia (UIP); increased MMP-9 levels have been detected in BAL fluid and pulmonary tissues in patients with these conditions. Acute lung injury (ALI), which is a disease characterized by the disruption of the diffuse alveolar–capillary wall, the invasion of circulating inflammatory cells, and deficient oxygenation caused by respiratory syncytial virus (RSV) infection and other viral infections. Increased MMP-9 levels have been detected in respiratory secretions of children with ALI. Acute respiratory distress syndrome (ARDS), which is the most severe manifestation of ALI and is characterized by excessive systemic inflammation, an augmented permeability of the alveolar epithelial–endothelial capillary barrier, tissue damage, and acute respiratory failure. Increased MMP-9 levels have been detected in (i) the plasma of patients with ARDS, (ii) BAL fluid corresponding to the acute phase of ARDS, and (iii) the BAL fluid of patients with ARDS associated with severe trauma or septic shock. Mechanically ventilated infants with RSV-induced respiratory failure. Infection with coronavirus; increased MMP-9 levels have been detected in the circulation of COVID-19 patients with respiratory failure. | Implicated in a variety of illnesses, including COPD, emphysema, asthma (e.g., by enabling cell migration and airway inflammatory reaction, increased susceptibility to airway remodeling upon exposure to cigarette smoke resulting in thickening of airway walls, and bronchopulmonary dysplasia), ALI, CF, neutrophilic inflammation-mediated VILI (which is a severe lung injury associated with high-pressure mechanical ventilation), mechanically ventilated infants with RSV-infected lungs, and interstitial lung diseases (such as IPF-UIP and diffuse alveolar damage). Promotes eosinophil migration into the airways from asthmatic patients. Stimulates the degradation of the alveolar capillary barrier and inflammation, further promoting the migration of inflammatory cells (e.g., neutrophil recruitment through the generation of collagen fragments with chemotactic properties) and the damage of lung tissue. Associated with diffuse alveolar injury in hyperoxia-induced ALI in pigs. Linked to inflammation-induced tissue remodeling. Associated with markers of basement membrane disruption, in the acute phase of ARDS. Exacerbates lung tissue remodeling in rat lungs with hyperoxia. Implicated, when MMP-9/TIMP-1 ratio is less than 1, in the evolution of ARDS to fibroproliferation. Contributes to the disruption of alveolar epithelial basement membrane and increases fibroblast migration to alveolar spaces in the initial stage of lung fibrosis. Associated with respiratory failure in COVID-19 patients. |
| MMP-12 | COPD; increased MMP-12 levels have been detected in the BAL fluids, bronchial biopsy tissue, BAL cells, and sputum of patients with COPD. Emphysema; increased MMP-12 levels have been detected in dendritic and bronchial epithelial cells in human lung in response to cigarette smoke. | Participates in the pathogenesis of acute and chronic respiratory disorders (such as COPD, cigarette smoke-induced emphysema, and asthma) when they are induced in ASMC by inflammatory cytokines under the regulation of different mechanisms (such as PI3-K, JNK, ERK, and AP-1 signaling pathways). Partially mediates the IL-13-induced expression of MMP-2, MMP-9, MMP-13, and MMP-14, which results in the development of COPD-related features (such as the accumulation of macrophages, eosinophils, and neutrophils, thus leading to lung inflammation, alveolar enlargement, and increased lung volume). Involved in smoke-induced inflammation and emphysema (e.g., by causing endothelial activation mediated by the release of TNF-α from macrophages, degrading connective tissue, and facilitating neutrophil influx). Facilitates airway inflammation by promoting the migration of inflammatory cells (e.g., macrophages and monocytes) to inflammatory zones. Implicated in airway remodeling by degrading extracellular matrix proteins (e.g., fibronectin, elastin, laminin, type IV collagen, and gelatin) or mediating inflammatory cytokines to stimulate production of other MMPs (MMP-2, MMP-9, MMP-13, and MMP-14) in the lung. In animal models, it facilitates the development of inflammatory processes involving the recruitment of inflammatory cells, increases in cytokine and chemokine levels, and elevation of MMPs (e.g., MMP-2 and MMP-9) levels in lung samples and BAL fluids. |
| MMP-14 | COPD; increased MMP-14 levels have been detected in samples (e.g., BAL fluid from patients with emphysema) from COPD patients upon acrolein induction. | Implicated in COPD pathogenesis. |
| MMPsubsets | COPD; levels are elevated for (i) MMP-1 and MMP-2 derived from epithelial cells and macrophages, MMP-8 and MMP-9 derived from neutrophils, MMP-12, and MMP-14—all in the pulmonary tissues of COPD patients; (ii) MMP-1, MMP-2, MMP-8, and MMP-9 in the BAL fluid of patients with COPD; and (iii) MMP-2 and MMP-9 in the sputum of patients with COPD. Emphysema; levels are elevated for (i) MMP-1 and MMP-9 in the BAL fluid of patients with emphysema; (ii) MMP-8 and MMP-9 in alveolar macrophages from patients with emphysema; and (iii) MMP-9 and MMP-12 induced by cigarette smoke in human lung. Asthma; levels are elevated for (i) MMP-1, MMP-2, MMP-3, MMP-8, and MMP-9 in the sputum and BAL of asthma patients; (ii) MMP-2, MMP-3, and MMP-14 (and also TIMP-1 and TIMP-2) in the ASMC of patients with bronchial asthma; and (iii) MMP-2 and MMP-12 in the bronchial epithelial, smooth muscle and submucosal glandular cells, BAL fluid, and sputum of subjects with bronchiectasis and asthma. IPF; levels are elevated for (i) MMP-3, MMP-7, MMP-8, and MMP-9 in the lavage fluid of patients with IPF. ALI; levels are elevated for (i) MMP-2, MMP-3, MMP-8, MMP-9, MMP-11, and MMP-12, generated mainly by neutrophil and macrophage cells in the lung secretions of pediatric patients with ALI. ARDS; levels are elevated for (i) MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, MMP-12, and MMP-13 in patients with ARDS; (ii) MMP-2 and MMP-9 in the lung of newborns with ARDS; and (iii) MMP-8 and MMP-9, and to a limited extent for MMP-3, MMP-11, and MMP-12 in the lung secretions of children with ALI/ARDS. Pneumonia; levels are elevated for (i) MMP-8 and MMP-9 in the BAL fluid of patients with pneumonia acquired in hospitals. Ageing lungs; levels are elevated for (i) MMP-9 and MMP-12 (and also TIMP-1 and TIMP-2) in ageing lungs. By contrast, decreased MMP levels are reported for MMP-1 and MMP-2 in ageing lungs. Pulmonary fibrosis; levels are elevated for (i) MMP-2 and MMP-9 in BAL fluid and pulmonary tissue associated with pulmonary fibrosis; (ii) MMP-1 (largely found in bronchiolar and alveolar epithelial cells), MMP-2, MMP-7 (in the epithelium of lungs with IPF and fibrotic lung extracts), and MMP-9 in the lungs of patients with IPF; (iii) MMP-3 and MMP-12 in human alveolar macrophages under the effect of surfactant protein D; and (iv) MMP-2, MMP-8, and MMP-9 in the BAL fluid of patients with CF. Bronchiolitis obliterans, which is an irreversible condition characterized by obstructive airway remodeling. Levels are elevated for (i) MMP-8 and MMP-9 from neutrophils recruited into the lung (and also for TIMP-1) in patients with advanced bronchiolitis obliterans. | MMP-2 and MMP-9: Involved in the pathogenesis (e.g., excessive tissue remodeling) of COPD. MMP-1, MMP-8, and MMP-9: associated with the pathogenesis of emphysema. Predominantly MMP-9 and several other MMPs (such as MMP-1, MMP-2, MMP-7, MMP-8, MMP-12, and MMP-25): associated with asthma pathology. MMP-3, MMP-7, MMP-8, and MMP-9: implicated in pulmonary fibrosis, by facilitating the release of extracellular matrix-derived VEGF, resulting in abnormal capillary permeability and neoangiogenesis. MMP-1, MMP-2, MMP-7, and MMP-9 (and also TIMP-1 and TIMP-2): show an active role in IPF. MMP-8 and MMP-9: associated with the grade of systemic inflammation in patients with ventilator-associated pneumonia. MMP-9 and MMP-12 (and also TIMP-1 and TIMP-2): associated with deteriorated lung function during ageing. MMP-3 and MMP-9: associated with pathogenic roles (e.g., interstitial matrix remodeling, rupture of the basement membranes) in ALI. MMP-2, MMP-9, and MMP-12: mediate structural lung damage (alveolar type I cell injury, pulmonary compliance variations, respiratory failure, and death) in mouse models (e.g., hyperoxia-induced lung disease). MMP-1, MMP-3, and MMP-12: involved in lung innate immune system; pulmonary surfactant proteins induces the expression of these enzymes in human alveolar macrophages. Activated macrophages in the lung clear offending pathogens (viruses, bacteria, and fungi) that enter the airway. MMP-9 and MMP-12: implicated in the effects of IL-1β in lungs of asthmatic patients; in the lungs of adult mice, IL-1β contributes to pulmonary inflammation (e.g., macrophage and neutrophil infiltration) and fibrosis in the airway. MMP-2, MMP-8, MMP-9 (and also TIMP-1) and sometimes MMP-1 and MMP-3: involved in neutrophil-mediated inflammation, extensive tissue remodeling, and the loss of the intercellular junctions and alveolar–capillary barrier that occur in ARDS. These roles are also valid for MMP-3, MMP-7, and MMP-12 in animal models. MMP-1 and/or MMP-3, together with MMP-2, MMP-8, and MMP-9 (and also TIMP-1): associated with more severe disease progression (i.e., higher indices of disease harshness, widespread multiorgan failure, and more mortality) in patients with ALI/ARDS. MMP-2 and MMP-9: cause an acute injury pattern in rats treated with bleomycin MMP-2, MMP-8, and MMP-9: play a role in the pathogenesis of ventilator-provoked lung damage. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardy, E.; Fernandez-Patron, C. Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis. Biomolecules 2021, 11, 390. https://doi.org/10.3390/biom11030390
Hardy E, Fernandez-Patron C. Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis. Biomolecules. 2021; 11(3):390. https://doi.org/10.3390/biom11030390
Chicago/Turabian StyleHardy, Eugenio, and Carlos Fernandez-Patron. 2021. "Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis" Biomolecules 11, no. 3: 390. https://doi.org/10.3390/biom11030390
APA StyleHardy, E., & Fernandez-Patron, C. (2021). Targeting MMP-Regulation of Inflammation to Increase Metabolic Tolerance to COVID-19 Pathologies: A Hypothesis. Biomolecules, 11(3), 390. https://doi.org/10.3390/biom11030390