
Infarct in the Heart: What’s MMP-9 Got to Do with It?

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
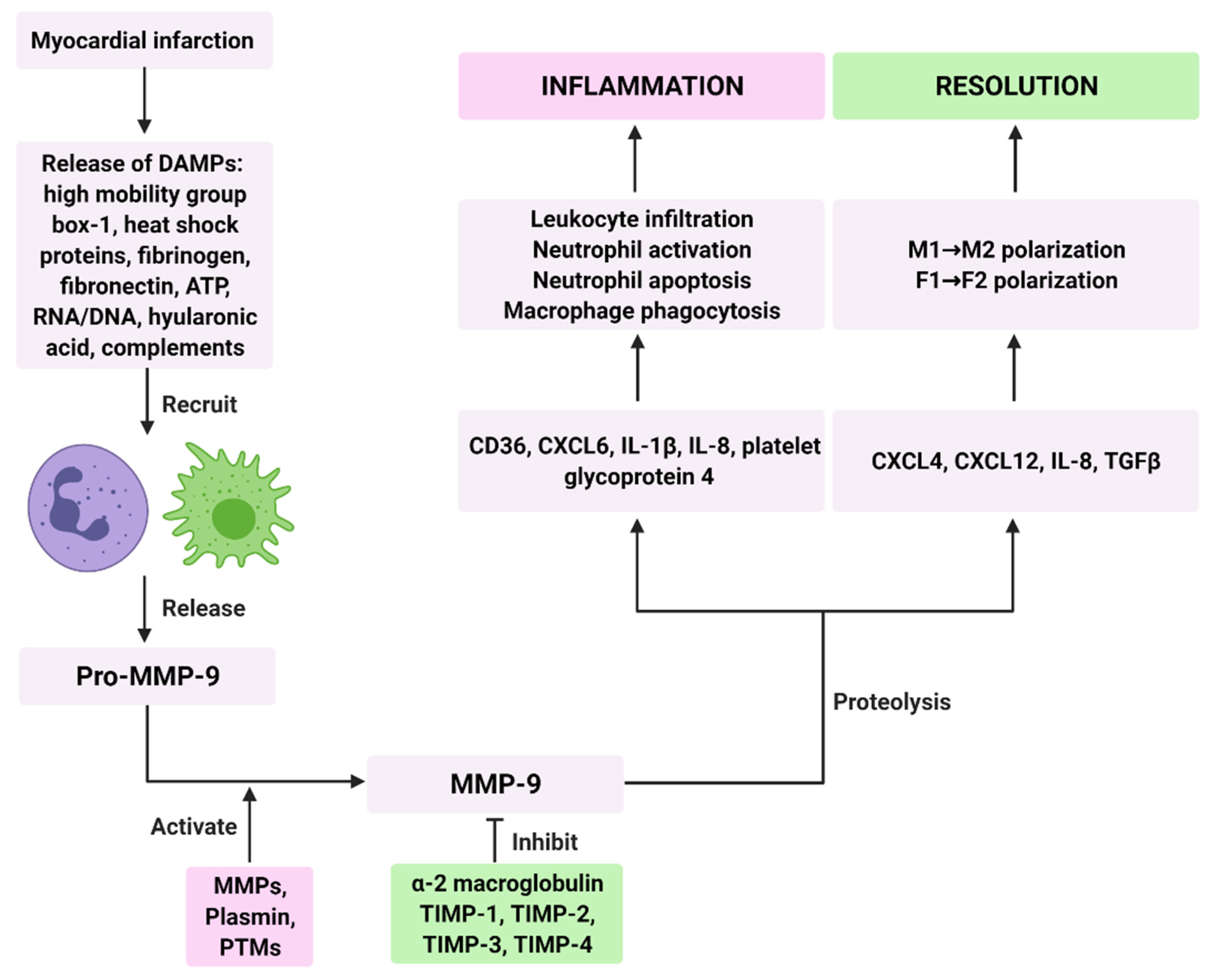
2. Regulation of MMP-9 Activity
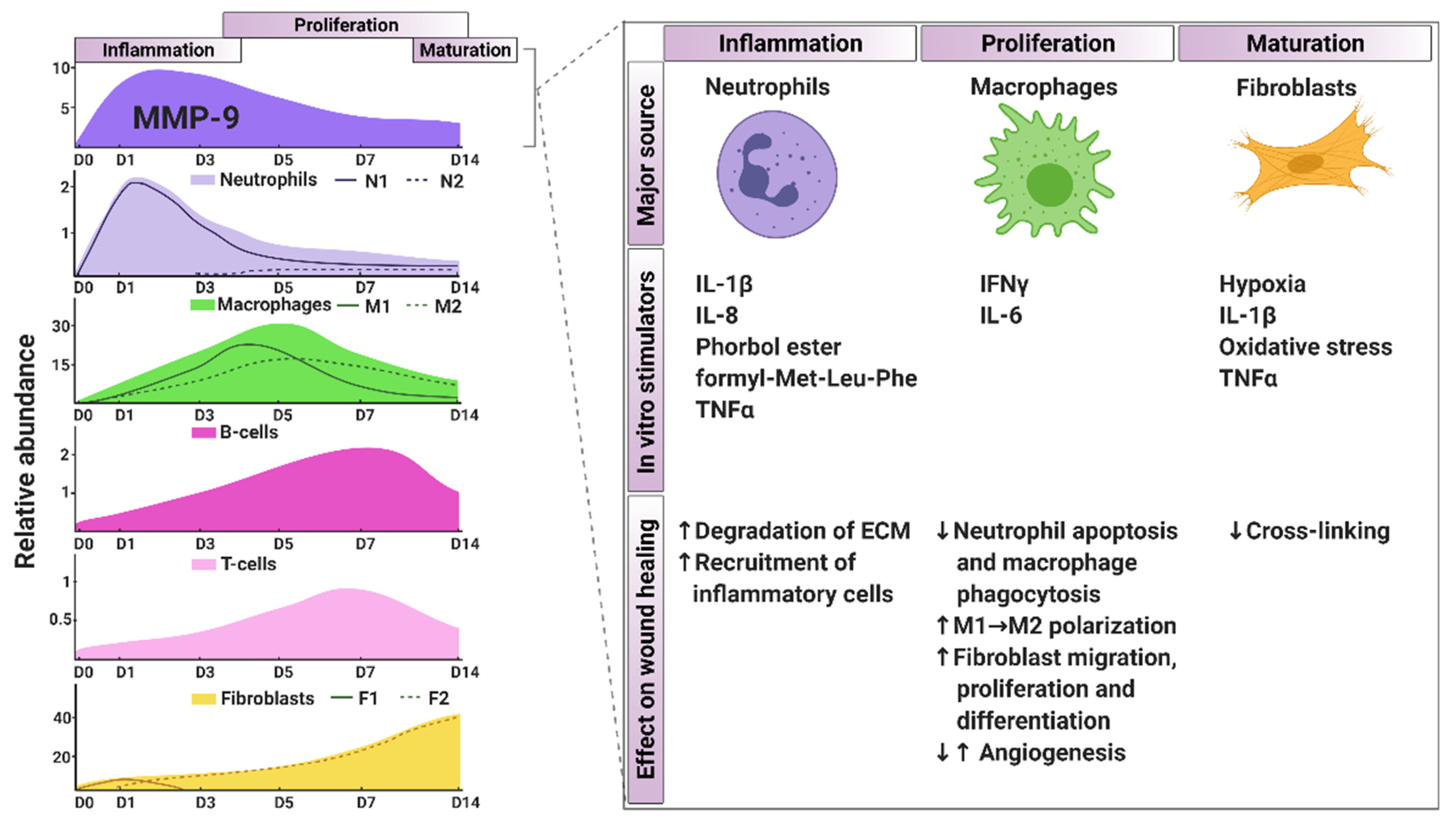
3. MMP-9 Signaling in the Inflammatory Phase
4. MMP-9 Signaling in the Proliferation Phase
5. MMP-9 Signaling in the Maturation Phase
6. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Ferrini, A.; Stevens, M.M.; Sattler, S.; Rosenthal, N. Toward Regeneration of the Heart: Bioengineering Strategies for Immunomodulation. Front. Cardiovasc. Med. 2019, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Kloner, R.A.; Brown, D.A.; Csete, M.; Dai, W.; Downey, J.M.; Gottlieb, R.A.; Hale, S.L.; Shi, J. New and revisited approaches to preserving the reperfused myocardium. Nat. Rev. Cardiol. 2017, 14, 679–693. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Clarke, S.A.; Richardson, W.J.; Holmes, J.W. Modifying the mechanics of healing infarcts: Is better the enemy of good? J. Mol. Cell. Cardiol. 2016, 93, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, W.T.; Zhang, Z.M.; Chang, P.P.; Rosamond, W.D.; Kitzman, D.W.; Wagenknecht, L.E.; Soliman, E.Z. Silent Myocardial Infarction and Long-Term Risk of Heart Failure: The ARIC Study. J. Am. Coll. Cardiol. 2018, 71, 1–8. [Google Scholar] [CrossRef]
- Yuan, X.; Braun, T. Multimodal Regulation of Cardiac Myocyte Proliferation. Circ. Res. 2017, 121, 293–309. [Google Scholar] [CrossRef]
- Mouton, A.J.; Rivera, O.J.; Lindsey, M.L. Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H71–H79. [Google Scholar] [CrossRef]
- Nahrendorf, M. Imaging of infarct healing predicts left ventricular remodeling and evolution of heart failure: Focus on protease activity. Circ. Cardiovasc. Imaging 2011, 4, 351–353. [Google Scholar] [CrossRef] [Green Version]
- White, H.D.; Norris, R.M.; Brown, M.A.; Brandt, P.W.; Whitlock, R.M.; Wild, C.J. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation 1987, 76, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Orn, S.; Manhenke, C.; Anand, I.S.; Squire, I.; Nagel, E.; Edvardsen, T.; Dickstein, K. Effect of left ventricular scar size, location, and transmurality on left ventricular remodeling with healed myocardial infarction. Am. J. Cardiol. 2007, 99, 1109–1114. [Google Scholar] [CrossRef]
- Miura, T.; Miki, T. Limitation of myocardial infarct size in the clinical setting: Current status and challenges in translating animal experiments into clinical therapy. Basic Res. Cardiol. 2008, 103, 501–513. [Google Scholar] [CrossRef]
- Lambert, L.; Brown, K.; Segal, E.; Brophy, J.; Rodes-Cabau, J.; Bogaty, P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA 2010, 303, 2148–2155. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Kain, V.; Prabhu, S.D.; Halade, G.V. Inflammation revisited: Inflammation versus resolution of inflammation following myocardial infarction. Basic Res. Cardiol. 2014, 109, 444. [Google Scholar] [CrossRef]
- Panizzi, P.; Swirski, F.K.; Figueiredo, J.L.; Waterman, P.; Sosnovik, D.E.; Aikawa, E.; Libby, P.; Pittet, M.; Weissleder, R.; Nahrendorf, M. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J. Am. Coll. Cardiol. 2010, 55, 1629–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamilpa, R.; Lindsey, M.L. Extracellular matrix turnover and signaling during cardiac remodeling following MI: Causes and consequences. J. Mol. Cell. Cardiol. 2010, 48, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, A.R.; Moore, E.T.; Daseke, M.J., 2nd; Valerio, F.M.; Flynn, E.R.; Lindsey, M.L. The compendium of matrix metalloproteinase expression in the left ventricle of mice following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H706–H714. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.P.; Jung, M.; Lindsey, M.L. MMP-9 signaling in the left ventricle following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H190–H198. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat. Rev. Cardiol. 2018, 15, 471–479. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar]
- Halade, G.V.; Jin, Y.F.; Lindsey, M.L. Matrix metalloproteinase (MMP)-9: A proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol. Ther. 2013, 139, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.J.; Zhao, Q.; Qu, H.J.; Li, X.M.; Chen, Q.J.; Liu, F.; Chen, B.D.; Yang, Y.N. Usefulness of plasma matrix metalloproteinase-9 levels in prediction of in-hospital mortality in patients who received emergent percutaneous coronary artery intervention following myocardial infarction. Oncotarget 2017, 8, 105809–105818. [Google Scholar] [CrossRef] [Green Version]
- Somuncu, M.U.; Pusuroglu, H.; Karakurt, H.; Bolat, İ.; Karakurt, S.T.; Demir, A.R.; Isıksacan, N.; Akgul, O.; Surgit, O. The prognostic value of elevated matrix metalloproteinase-9 in patients undergoing primary percutaneous coronary intervention for ST-elevation myocardial infarction: A two-year prospective study. Rev. Port. Cardiol. 2020, 39, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Squire, I.B.; Evans, J.; Ng, L.L.; Loftus, I.M.; Thompson, M.M. Plasma MMP-9 and MMP-2 following acute myocardial infarction in man: Correlation with echocardiographic and neurohumoral parameters of left ventricular dysfunction. J. Card. Fail. 2004, 10, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Cockerill, G.; Ng, L.L.; Thompson, M.; Khan, S.; Samani, N.J.; Squire, I.B. Plasma matrix metalloproteinase-9 and left ventricular remodelling after acute myocardial infarction in man: A prospective cohort study. Eur. Heart J. 2007, 28, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Van den Borne, S.W.; Cleutjens, J.P.; Hanemaaijer, R.; Creemers, E.E.; Smits, J.F.; Daemen, M.J.; Blankesteijn, W.M. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc. Pathol. 2009, 18, 37–43. [Google Scholar] [CrossRef]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Zee, J.M.; Patel, K.D. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: Novel pathways for tertiary granule release. J. Leukoc. Biol. 2006, 79, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Puhl, S.L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Deleon-Pennell, K.Y.; Altara, R.; Yabluchanskiy, A.; Modesti, A.; Lindsey, M.L. The circular relationship between matrix metalloproteinase-9 and inflammation following myocardial infarction. IUBMB Life 2015, 67, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Papazafiropoulou, A.; Tentolouris, N. Matrix metalloproteinases and cardiovascular diseases. Hippokratia 2009, 13, 76–82. [Google Scholar]
- O’Sullivan, S.; Medina, C.; Ledwidge, M.; Radomski, M.W.; Gilmer, J.F. Nitric oxide-matrix metaloproteinase-9 interactions: Biological and pharmacological significance—NO and MMP-9 interactions. Biochim. Biophys. Acta 2014, 1843, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Boon, L.; Ugarte-Berzal, E.; Vandooren, J.; Opdenakker, G. Glycosylation of matrix metalloproteases and tissue inhibitors: Present state, challenges and opportunities. Biochem. J. 2016, 473, 1471–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybakowski, J.K. Matrix Metalloproteinase-9 (MMP9)-A Mediating Enzyme in Cardiovascular Disease, Cancer, and Neuropsychiatric Disorders. Cardiovasc. Psychiatry Neurol. 2009, 2009, 904836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Shastri, V.P. Matrix-metalloproteinase-9 is cleaved and activated by cathepsin K. BMC Res. Notes 2015, 8, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, M.; Chvyrkova, I.; Bernardo, M.M.; Hernandez-Barrantes, S.; Fridman, R. Pro-MMP-9 activation by the MT1-MMP/MMP-2 axis and MMP-3: Role of TIMP-2 and plasma membranes. Biochem. Biophys. Res. Commun. 2003, 308, 386–395. [Google Scholar] [CrossRef]
- Fridman, R.; Toth, M.; Peña, D.; Mobashery, S. Activation of progelatinase B (MMP-9) by gelatinase A (MMP-2). Cancer Res. 1995, 55, 2548–2555. [Google Scholar]
- Ramos-DeSimone, N.; Hahn-Dantona, E.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 1999, 274, 13066–13076. [Google Scholar] [CrossRef] [Green Version]
- von Bredow, D.C.; Cress, A.E.; Howard, E.W.; Bowden, G.T.; Nagle, R.B. Activation of gelatinase-tissue-inhibitors-of-metalloproteinase complexes by matrilysin. Biochem. J. 1998, 331 Pt 3, 965–972. [Google Scholar] [CrossRef]
- Knäuper, V.; Smith, B.; López-Otin, C.; Murphy, G. Activation of progelatinase B (proMMP-9) by active collagenase-3 (MMP-13). Eur. J. Biochem. 1997, 248, 369–373. [Google Scholar] [CrossRef]
- Davis, G.E.; Pintar Allen, K.A.; Salazar, R.; Maxwell, S.A. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J. Cell Sci. 2001, 114 Pt 5, 917–930. [Google Scholar]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology 2013, 28, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Serifova, X.; Ugarte-Berzal, E.; Opdenakker, G.; Vandooren, J. Homotrimeric MMP-9 is an active hitchhiker on alpha-2-macroglobulin partially escaping protease inhibition and internalization through LRP-1. Cell. Mol. Life Sci. 2020, 77, 3013–3026. [Google Scholar] [CrossRef]
- Rehman, A.A.; Ahsan, H.; Khan, F.H. α-2-Macroglobulin: A physiological guardian. J. Cell. Physiol. 2013, 228, 1665–1675. [Google Scholar] [CrossRef]
- Duellman, T.; Burnett, J.; Yang, J. Functional Roles of N-Linked Glycosylation of Human Matrix Metalloproteinase 9. Traffic 2015, 16, 1108–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn-Dantona, E.; Ruiz, J.F.; Bornstein, P.; Strickland, D.K. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J. Biol. Chem. 2001, 276, 15498–15503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, W.; Fernandez-Catalan, C.; Nagase, H.; Maskos, K. Endoproteinase-protein inhibitor interactions. Apmis 1999, 107, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur. J. Cell Biol. 1997, 74, 111–122. [Google Scholar]
- Roten, L.; Nemoto, S.; Simsic, J.; Coker, M.L.; Rao, V.; Baicu, S.; Defreyte, G.; Soloway, P.J.; Zile, M.R.; Spinale, F.G. Effects of gene deletion of the tissue inhibitor of the matrix metalloproteinase-type 1 (TIMP-1) on left ventricular geometry and function in mice. J. Mol. Cell. Cardiol. 2000, 32, 109–120. [Google Scholar] [CrossRef]
- Ikonomidis, J.S.; Hendrick, J.W.; Parkhurst, A.M.; Herron, A.R.; Escobar, P.G.; Dowdy, K.B.; Stroud, R.E.; Hapke, E.; Zile, M.R.; Spinale, F.G. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: Effects of exogenous MMP inhibition. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H149–H158. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt 11, 2015–2025. [Google Scholar] [CrossRef]
- Kandalam, V.; Basu, R.; Abraham, T.; Wang, X.; Soloway, P.D.; Jaworski, D.M.; Oudit, G.Y.; Kassiri, Z. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ. Res. 2010, 106, 796–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoutte, D.; Schellings, M.; Pinto, Y.; Heymans, S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: A temporal and spatial window. Cardiovasc. Res. 2006, 69, 604–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roderfeld, M.; Graf, J.; Giese, B.; Salguero-Palacios, R.; Tschuschner, A.; Müller-Newen, G.; Roeb, E. Latent MMP-9 is bound to TIMP-1 before secretion. Biol. Chem. 2007, 388, 1227–1234. [Google Scholar] [CrossRef]
- Gearing, A.J.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L.; et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Butler, G.S.; Gong, J.H.; Bendall, L.; Power, C.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 2001, 276, 43503–43508. [Google Scholar] [CrossRef] [Green Version]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar]
- Van Den Steen, P.E.; Wuyts, A.; Husson, S.J.; Proost, P.; Van Damme, J.; Opdenakker, G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur. J. Biochem. 2003, 270, 3739–3749. [Google Scholar] [CrossRef]
- Meschiari, C.A.; Jung, M.; Iyer, R.P.; Yabluchanskiy, A.; Toba, H.; Garrett, M.R.; Lindsey, M.L. Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H224–H235. [Google Scholar] [CrossRef]
- Iyer, R.P.; de Castro Bras, L.E.; Patterson, N.L.; Bhowmick, M.; Flynn, E.R.; Asher, M.; Cannon, P.L.; Deleon-Pennell, K.Y.; Fields, G.B.; Lindsey, M.L. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J. Mol. Cell. Cardiol. 2016, 100, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabluchanskiy, A.; Ma, Y.; DeLeon-Pennell, K.Y.; Altara, R.; Halade, G.V.; Voorhees, A.P.; Nguyen, N.T.; Jin, Y.F.; Winniford, M.D.; Hall, M.E.; et al. Myocardial Infarction Superimposed on Aging: MMP-9 Deletion Promotes M2 Macrophage Polarization. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Iyer, R.P.; Zamilpa, R.; Yabluchanskiy, A.; DeLeon-Pennell, K.Y.; Hall, M.E.; Kaplan, A.; Zouein, F.A.; Bratton, D.; Flynn, E.R.; et al. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J. Am. Coll. Cardiol. 2015, 66, 1364–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Z.; Peng, Q.; Liu, Y.Y.; Zhang, W.; Wu, L.; Wang, X.; Luo, F. Tumor refractoriness to endostatin anti-angiogenesis is associated with the recruitment of CD11b+Gr1+ myeloid cells and inflammatory cytokines. Tumori 2013, 99, 723–733. [Google Scholar] [CrossRef]
- Ferreras, M.; Felbor, U.; Lenhard, T.; Olsen, B.R.; Delaissé, J. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett. 2000, 486, 247–251. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Zouein, F.A.; Tian, Y.; Padmanabhan Iyer, R.; de Castro Bras, L.E. Osteopontin is proteolytically processed by matrix metalloproteinase 9. Can. J. Physiol. Pharmacol. 2015, 93, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Furtado, M.B.; Rosenthal, N. The interstitium in cardiac repair: Role of the immune-stromal cell interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef]
- Liehn, E.A.; Postea, O.; Curaj, A.; Marx, N. Repair after myocardial infarction, between fantasy and reality: The role of chemokines. J. Am. Coll. Cardiol. 2011, 58, 2357–2362. [Google Scholar] [CrossRef] [Green Version]
- Bonvini, R.F.; Hendiri, T.; Camenzind, E. Inflammatory response post-myocardial infarction and reperfusion: A new therapeutic target? Eur. Heart J. Suppl. 2005, 7 (Suppl. I), I27–I36. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.; Ma, Y.; Li, A.; Shi, H.; Nie, S. Trimetazidine suppresses oxidative stress, inhibits MMP-2 and MMP-9 expression, and prevents cardiac rupture in mice with myocardial infarction. Cardiovasc. Ther. 2018, 36, e12460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Daseke, M.J., 2nd; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell Signal. 2021, 77, 109816. [Google Scholar] [CrossRef]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marom, B.; Rahat, M.A.; Lahat, N.; Weiss-Cerem, L.; Kinarty, A.; Bitterman, H. Native and fragmented fibronectin oppositely modulate monocyte secretion of MMP-9. J. Leukoc. Biol. 2007, 81, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; Ma, Y.; Ramirez, T.A.; Zhang, J.; Dai, Q.; Hensler, J.G.; Lopez, E.F.; Ghasemi, O.; Jin, Y.F.; Lindsey, M.L. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1830–H1842. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.F.; Han, H.C.; Berger, J.; Dai, Q.; Lindsey, M.L. Combining experimental and mathematical modeling to reveal mechanisms of macrophage-dependent left ventricular remodeling. BMC Syst. Biol. 2011, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.Y.; Cavasin, M.A.; Yang, F.; Liu, Y.H.; Yang, X.P. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004, 74, 1561–1572. [Google Scholar] [CrossRef]
- Ma, Y.; Yabluchanskiy, A.; Lindsey, M.L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, T.; Ma, Y.; Halade, G.V.; Zhang, J.; Lindsey, M.L.; Jin, Y.F. Mathematical modeling and stability analysis of macrophage activation in left ventricular remodeling post-myocardial infarction. BMC Genom. 2012, 13 (Suppl. S6), S21. [Google Scholar] [CrossRef] [Green Version]
- BioRender. Available online: https://biorender.com/ (accessed on 23 March 2021).
- Lindsey, M.; Wedin, K.; Brown, M.D.; Keller, C.; Evans, A.J.; Smolen, J.; Burns, A.R.; Rossen, R.D.; Michael, L.; Entman, M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001, 103, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Joffs, C.; Deschamps, A.M.; Davis, J.; Dowdy, K.; Hendrick, J.; Baicu, S.; Mukherjee, R.; Manhaini, M.; Spinale, F.G. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H987–H994. [Google Scholar] [CrossRef] [PubMed]
- Romanic, A.M.; Burns-Kurtis, C.L.; Gout, B.; Berrebi-Bertrand, I.; Ohlstein, E.H. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci. 2001, 68, 799–814. [Google Scholar] [CrossRef]
- Creemers, E.E.; Cleutjens, J.P.; Smits, J.F.; Daemen, M.J. Matrix metalloproteinase inhibition after myocardial infarction: A new approach to prevent heart failure? Circ. Res. 2001, 89, 201–210. [Google Scholar] [CrossRef] [Green Version]
- DeLeon-Pennell, K.Y.; Tian, Y.; Zhang, B.; Cates, C.A.; Iyer, R.P.; Cannon, P.; Shah, P.; Aiyetan, P.; Halade, G.V.; Ma, Y.; et al. CD36 Is a Matrix Metalloproteinase-9 Substrate That Stimulates Neutrophil Apoptosis and Removal During Cardiac Remodeling. Circ. Cardiovasc. Genet. 2016, 9, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Zamilpa, R.; Ibarra, J.; de Castro Bras, L.E.; Ramirez, T.A.; Nguyen, N.; Halade, G.V.; Zhang, J.; Dai, Q.; Dayah, T.; Chiao, Y.A.; et al. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J. Mol. Cell. Cardiol. 2012, 53, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Selejan, S.R.; Hewera, L.; Hohl, M.; Kazakov, A.; Ewen, S.; Kindermann, I.; Böhm, M.; Link, A. Suppressed MMP-9 Activity in Myocardial Infarction-Related Cardiogenic Shock Implies Diminished Rage Degradation. Shock 2017, 48, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Takeishi, Y.; Niizeki, T.; Suzuki, S.; Kitahara, T.; Sasaki, T.; Kubota, I. Soluble Receptor for advanced glycation end products (RAGE) is a prognostic factor for heart failure. J. Card. Fail. 2008, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varda-Bloom, N.; Leor, J.; Ohad, D.G.; Hasin, Y.; Amar, M.; Fixler, R.; Battler, A.; Eldar, M.; Hasin, D. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J. Mol. Cell. Cardiol. 2000, 32, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Pitts, C.; Clayton, J.; Domondon, M.; Troncoso, M.; Pippin, S.; DeLeon-Pennell, K.Y. CD8(+) T-cells negatively regulate inflammation post-myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H581–H596. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Rivera Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Luttun, A.; Nuyens, D.; Theilmeier, G.; Creemers, E.; Moons, L.; Dyspersin, G.D.; Cleutjens, J.P.M.; Shipley, M.; Angellilo, A.; et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999, 5, 1135–1142. [Google Scholar] [CrossRef]
- He, B.J.; Joiner, M.L.; Singh, M.V.; Luczak, E.D.; Swaminathan, P.D.; Koval, O.M.; Kutschke, W.; Allamargot, C.; Yang, J.; Guan, X.; et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 2011, 17, 1610–1618. [Google Scholar] [CrossRef] [Green Version]
- Barlaka, E.; Görbe, A.; Gáspár, R.; Pálóczi, J.; Ferdinandy, P.; Lazou, A. Activation of PPARβ/δ protects cardiac myocytes from oxidative stress-induced apoptosis by suppressing generation of reactive oxygen/nitrogen species and expression of matrix metalloproteinases. Pharmacol. Res. 2015, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Jourdan-Lesaux, C.; Zhang, J.; Lindsey, M.L. Extracellular matrix roles during cardiac repair. Life Sci. 2010, 87, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell. Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef]
- Poon, E.N.; Luo, X.L.; Webb, S.E.; Yan, B.; Zhao, R.; Wu, S.C.M.; Yang, Y.; Zhang, P.; Bai, H.; Shao, J.; et al. The cell surface marker CD36 selectively identifies matured, mitochondria-rich hPSC-cardiomyocytes. Cell Res. 2020, 30, 626–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chiao, Y.A.; Clark, R.; Flynn, E.R.; Yabluchanskiy, A.; Ghasemi, O.; Zouein, F.; Lindsey, M.L.; Jin, Y.F. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 2015, 106, 421–431. [Google Scholar] [CrossRef]
- Spinale, F.G.; Frangogiannis, N.G.; Hinz, B.; Holmes, J.W.; Kassiri, Z.; Lindsey, M.L. Crossing into the Next Frontier of Cardiac Extracellular Matrix Research. Circ. Res. 2016, 119, 1040–1045. [Google Scholar] [CrossRef]
- Iyer, R.P.; de Castro Bras, L.E.; Jin, Y.F.; Lindsey, M.L. Translating Koch’s postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: Cardiac metalloproteinase actions (CarMA) postulates. Circ. Res. 2014, 114, 860–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, F.; Chen, J.; Shen, X.; Deng, Y.; Xu, L.; Yin, J.; Chen, H.; Teng, F.; Liu, X.; et al. Matrix metalloproteinase-9 induces cardiac fibroblast migration, collagen and cytokine secretion: Inhibition by salvianolic acid B from Salvia miltiorrhiza. Phytomedicine 2011, 19, 13–19. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Escobar, G.P.; Dobrucki, L.W.; Goshorn, D.K.; Bouges, S.; Mingoia, J.T.; McClister, D.M., Jr.; Su, H.; Gannon, J.; MacGillivray, C.; et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H232–H239. [Google Scholar] [CrossRef] [Green Version]
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol. 2019, 75–76, 170–189. [Google Scholar] [CrossRef]
- Johnson, C.; Sung, H.J.; Lessner, S.M.; Fini, M.E.; Galis, Z.S. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: Potential role in capillary branching. Circ. Res. 2004, 94, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Guo, M.; Zeng, W.; Wang, Y.; Yang, L.; Pang, X.; Li, H.; Suo, Y.; Jiang, X.; Yu, C. Matrix metalloproteinase 9 secreted by hypoxia cardiac fibroblasts triggers cardiac stem cell migration in vitro. Stem Cells Int. 2015, 2015, 836390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virag, J.I.; Murry, C.E. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am. J. Pathol. 2003, 163, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, A.P.; DeLeon-Pennell, K.Y.; Ma, Y.; Halade, G.V.; Yabluchanskiy, A.; Iyer, R.P.; Flynn, E.; Cates, C.A.; Lindsey, M.L.; Han, H.C. Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J. Mol. Cell. Cardiol. 2015, 85, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, T.A.; Iyer, R.P.; Ghasemi, O.; Lopez, E.F.; Levin, D.B.; Zhang, J.; Zamilpa, R.; Chou, Y.M.; Jin, Y.F.; Lindsey, M.L. Aliskiren and valsartan mediate left ventricular remodeling post-myocardial infarction in mice through MMP-9 effects. J. Mol. Cell. Cardiol. 2014, 72, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Ueland, T.; Holter, J.C.; Holten, A.R.; Müller, K.E.; Lind, A.; Bekken, G.K.; Dudman, S.; Aukrust, P.; Dyrhol-Riise, A.M.; Heggelund, L. Distinct and early increase in circulating MMP-9 in COVID-19 patients with respiratory failure. J. Infect. 2020, 81, e41–e43. [Google Scholar] [CrossRef]
- Seizer, P.; Klingel, K.; Sauter, M.; Westermann, D.; Ochmann, C.; Schönberger, T.; Schleicher, R.; Stellos, K.; Schmidt, E.M.; Borst, O.; et al. Cyclophilin A affects inflammation, virus elimination and myocardial fibrosis in coxsackievirus B3-induced myocarditis. J. Mol. Cell. Cardiol. 2012, 53, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Marchant, D.; Walker, E.K.; Luo, Z.; Zhang, J.; Yanagawa, B.; Rahmani, M.; Cox, J.; Overall, C.; Senior, R.M.; et al. Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice. Circulation 2008, 117, 1574–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, R.; Gundry, R.L.; Brett-Major, D.M.; Mahr, C.; Thiele, G.M.; Lindsey, M.L.; Anderson, D.R. COVID-19 and cardiovascular disease: What we know, what we think we know, and what we need to know. J. Mol. Cell. Cardiol. 2020, 144, 12–14. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ECM Parent Protein | ECM-Fragment | Effect on Cardiac Wound Healing | Reference |
|---|---|---|---|
| Collagen I | C-1158/59 | Stimulates neovascularization | [65] |
| Collagen IV | Tumstatin | Inhibits angiogenesis | [66] |
| Collagen XVIII | Endostatin | Inhibits angiogenesis | [67,68] |
| Osteopontin (OPN) | OPN-p151 OPN-p152 | Increases fibroblast migration and wound healing | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becirovic-Agic, M.; Chalise, U.; Daseke, M.J., II; Konfrst, S.; Salomon, J.D.; Mishra, P.K.; Lindsey, M.L. Infarct in the Heart: What’s MMP-9 Got to Do with It? Biomolecules 2021, 11, 491. https://doi.org/10.3390/biom11040491
Becirovic-Agic M, Chalise U, Daseke MJ II, Konfrst S, Salomon JD, Mishra PK, Lindsey ML. Infarct in the Heart: What’s MMP-9 Got to Do with It? Biomolecules. 2021; 11(4):491. https://doi.org/10.3390/biom11040491
Chicago/Turabian StyleBecirovic-Agic, Mediha, Upendra Chalise, Michael J. Daseke, II, Shelby Konfrst, Jeffrey D. Salomon, Paras K. Mishra, and Merry L. Lindsey. 2021. "Infarct in the Heart: What’s MMP-9 Got to Do with It?" Biomolecules 11, no. 4: 491. https://doi.org/10.3390/biom11040491
APA StyleBecirovic-Agic, M., Chalise, U., Daseke, M. J., II, Konfrst, S., Salomon, J. D., Mishra, P. K., & Lindsey, M. L. (2021). Infarct in the Heart: What’s MMP-9 Got to Do with It? Biomolecules, 11(4), 491. https://doi.org/10.3390/biom11040491