
The Ablation of VEGFR-1 Signaling Promotes Pressure Overload-Induced Cardiac Dysfunction and Sudden Death

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Angiotensin II Infusion
2.2. Echocardiography and Electrocardiography Recording
2.3. Immunohistochemistry
2.4. Western Blotting
2.5. RNA Isolation and RT-qPCR
2.6. RNA Sequencing
2.7. Data Availability
2.8. Statistical Analysis
3. Results
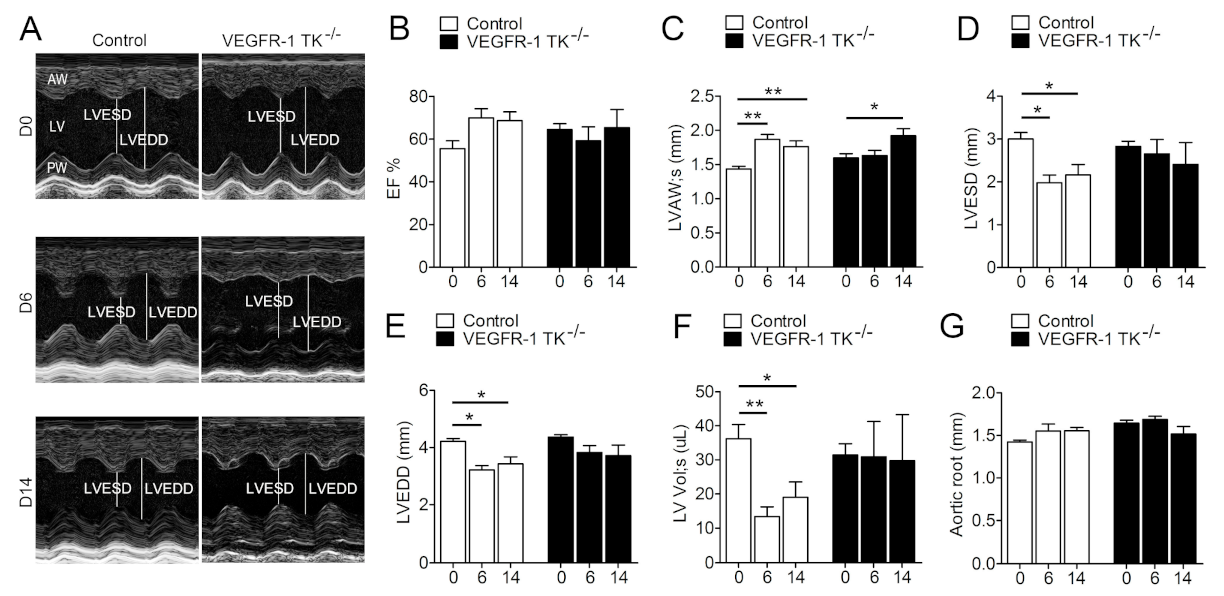
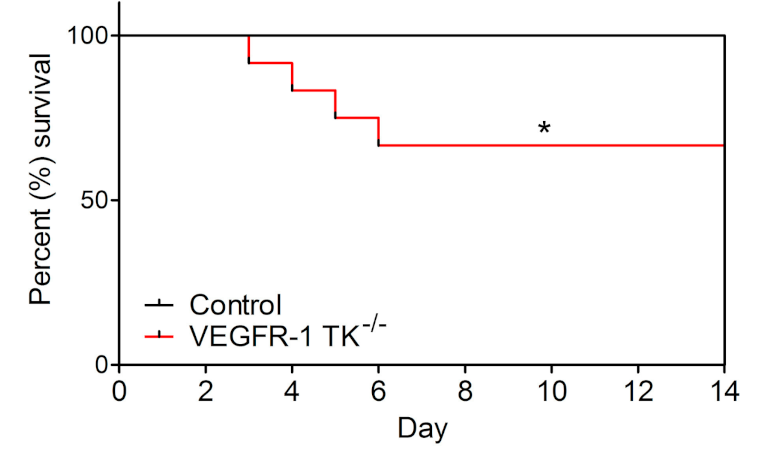
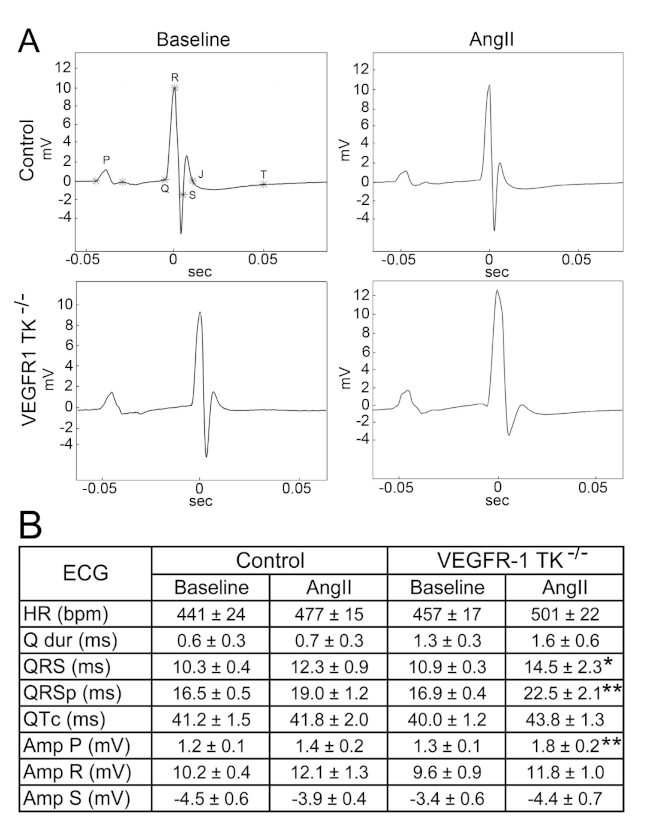
3.1. Deletion of the VEGFR-1 Signaling Halts the Early Development of Myocardial Compensatory Hypertrophy and Leads to Cardiac Dysfunction
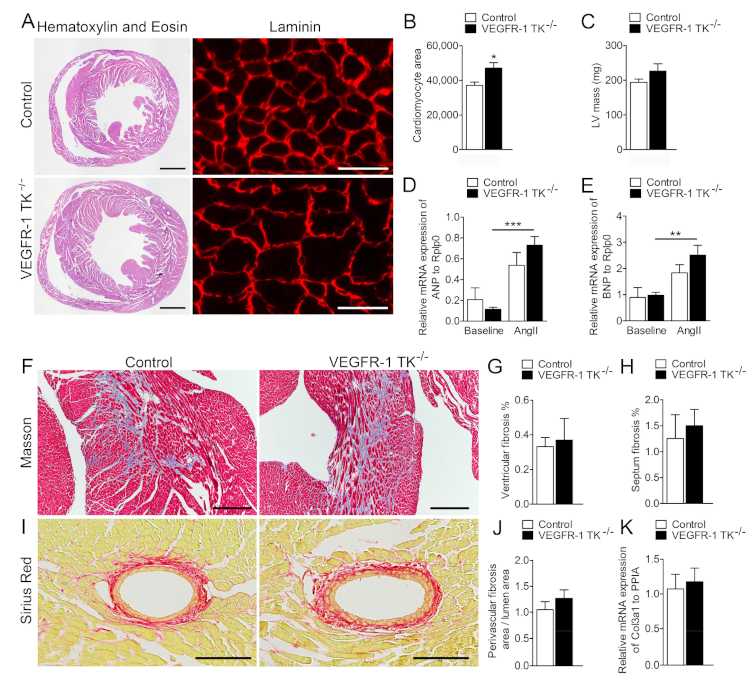
3.2. VEGFR-1 Tyrosine Kinase Deficiency Results in a Delayed but more Robust Development of Cardiomyocyte Hypertrophy
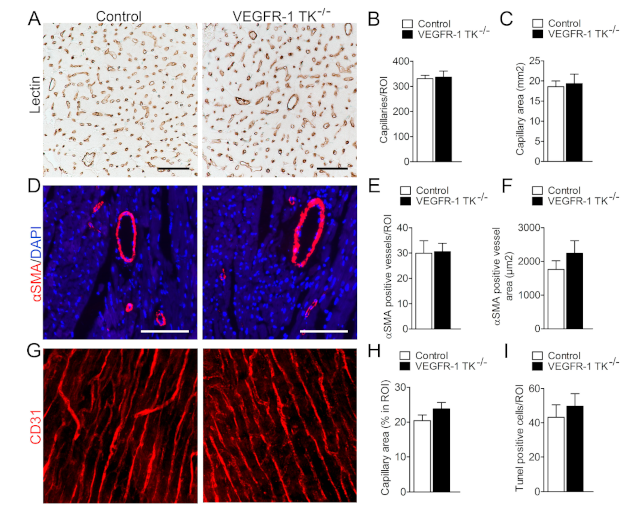
3.3. Deletion of VEGFR-1 Signaling Has no Effect on Angiogenesis in the Myocardium
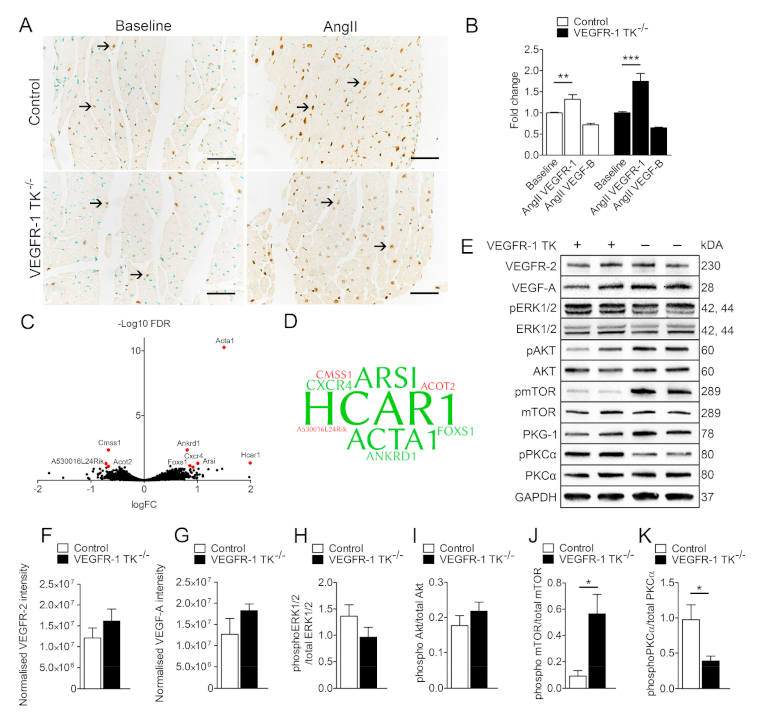
3.4. VEGFR-1 Levels Are Increased in the Myocardium during LVH
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef]
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition From Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail. 2017, 5, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Huusko, J.; Kurki, S.; Toppila, I.; Purmonen, T.; Lassenius, M.; Gullberg, E.; Wirta, S.B.; Ukkonen, H. Heart failure in Finland: Clinical characteristics, mortality, and healthcare resource use. ESC Heart Fail. 2019, 6, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Kerkelä, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2015, 4, e002423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huusko, J.; Merentie, M.; Dijkstra, M.H.; Ryhänen, M.-M.; Karvinen, H.; Rissanen, T.T.; Vanwildemeersch, M.; Hedman, M.; Lipponen, J.; Heinonen, S.E.; et al. The effects of VEGF-R1 and VEGF-R2 ligands on angiogenic responses and left ventricular function in mice. Cardiovasc. Res. 2010, 86, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lähteenvuo, J.E.; Lähteenvuo, M.T.; Kivelä, A.; Rosenlew, C.; Falkevall, A.; Klar, J.; Heikura, T.; Rissanen, T.T.; Vähäkangas, E.; Korpisalo, P.; et al. Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1- and neuropilin receptor-1-dependent mechanisms. Circulation 2009, 119, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Serpi, R.; Tolonen, A.-M.; Huusko, J.; Rysä, J.; Tenhunen, O.; Ylä-Herttuala, S.; Ruskoaho, H. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc. Res. 2011, 89, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Robciuc, M.R.; Kivelä, R.; Williams, I.M.; de Boer, J.F.; van Dijk, T.H.; Elamaa, H.; Tigistu-Sahle, F.; Molotkov, D.; Leppänen, V.-M.; Käkelä, R.; et al. VEGFB/VEGFR1-Induced Expansion of Adipose Vasculature Counteracts Obesity and Related Metabolic Complications. Cell Metab. 2016, 23, 712–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, V.C.; Duan, L.-J.; Cronin, C.; Liang, B.T.; Fong, G.-H. Elevated vascular endothelial growth factor receptor-2 abundance contributes to increased angiogenesis in vascular endothelial growth factor receptor-1-deficient mice. Circulation 2012, 126, 741–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivelä, R.; Bry, M.; Robciuc, M.R.; Räsänen, M.; Taavitsainen, M.; Silvola, J.M.U.; Saraste, A.; Hulmi, J.J.; Anisimov, A.; Mäyränpää, M.I.; et al. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol. Med. 2014, 6, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef] [Green Version]
- de Vries, C.; Escobedo, J.A.; Ueno, H.; Houck, K.; Ferrara, N.; Williams, L.T. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Huusko, J.; Lottonen, L.; Merentie, M.; Gurzeler, E.; Anisimov, A.; Miyanohara, A.; Alitalo, K.; Tavi, P.; Ylä-Herttuala, S. AAV9-mediated VEGF-B gene transfer improves systolic function in progressive left ventricular hypertrophy. Mol. Ther. 2012, 20, 2212–2221. [Google Scholar] [CrossRef] [Green Version]
- Merentie, M.; Lipponen, J.A.; Hedman, M.; Hedman, A.; Hartikainen, J.; Huusko, J.; Lottonen-Raikaslehto, L.; Parviainen, V.; Laidinen, S.; Karjalainen, P.A.; et al. Mouse ECG findings in aging, with conduction system affecting drugs and in cardiac pathologies: Development and validation of ECG analysis algorithm in mice. Physiol. Rep. 2015, 3, e12639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abràmoff, M.D.; Hospitals, I.; Magalhães, P.J.; Abràmoff, M. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar] [CrossRef]
- Mutikainen, M.; Tuomainen, T.; Naumenko, N.; Huusko, J.; Smirin, B.; Laidinen, S.; Kokki, K.; Hynynen, H.; Ylä-Herttuala, S.; Heinäniemi, M.; et al. Peroxisome proliferator-activated receptor-γ coactivator 1 α1 induces a cardiac excitation-contraction coupling phenotype without metabolic remodelling. J. Physiol. 2016, 594, 7049–7071. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, R.; Hemanthakumar, K.A.; Vaparanta, K.; Robciuc, M.; Izumiya, Y.; Kidoya, H.; Takakura, N.; Peng, X.; Sawyer, D.B.; Elenius, K.; et al. Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling. Circulation 2019, 139, 2570–2584. [Google Scholar] [CrossRef]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef]
- Wallenius, K.; Thalén, P.; Björkman, J.-A.; Johannesson, P.; Wiseman, J.; Böttcher, G.; Fjellström, O.; Oakes, N.D. Involvement of the metabolic sensor GPR81 in cardiovascular control. JCI Insight 2017, 2, e92564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suurmeijer, A.J.; Clément, S.; Francesconi, A.; Bocchi, L.; Angelini, A.; Van Veldhuisen, D.J.; Spagnoli, L.G.; Gabbiani, G.; Orlandi, A. α-Actin isoform distribution in normal and failing human heart: A morphological, morphometric, and biochemical study. J. Pathol. 2003, 199, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Stilli, D.; Bocchi, L.; Berni, R.; Zaniboni, M.; Cacciani, F.; Chaponnier, C.; Musso, E.; Gabbiani, G.; Clément, S. Correlation of α-skeletal actin expression, ventricular fibrosis and heart function with the degree of pressure overload cardiac hypertrophy in rats. Exp. Physiol. 2006, 91, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.S.M.; Chen, Y.-T.; Wang, J.; Richards, A.M.; Liew, O.W. Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential. Int. J. Mol. Sci. 2017, 18, 1362. [Google Scholar] [CrossRef]
- Amano, H.; Kato, S.; Ito, Y.; Eshima, K.; Ogawa, F.; Takahashi, R.; Sekiguchi, K.; Tamaki, H.; Sakagami, H.; Shibuya, M.; et al. The Role of Vascular Endothelial Growth Factor Receptor-1 Signaling in the Recovery from Ischemia. PLoS ONE 2015, 10, e0131445. [Google Scholar] [CrossRef] [PubMed]
- Amano, H.; Mastui, Y.; Ito, Y.; Shibata, Y.; Betto, T.; Eshima, K.; Ogawa, F.; Satoh, Y.; Shibuya, M.; Majima, M. The role of vascular endothelial growth factor receptor 1 tyrosine kinase signaling in bleomycin-induced pulmonary fibrosis. Biomed. Pharmacother. 2019, 117, 109067. [Google Scholar] [CrossRef]
- Larocca, T.J.; Jeong, D.; Kohlbrenner, E.; Lee, A.; Chen, J.; Hajjar, R.J.; Tarzami, S.T. CXCR4 gene transfer prevents pressure overload induced heart failure. J. Mol. Cell. Cardiol. 2012, 53, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef]
- Braz, J.C.; Bueno, O.F.; De Windt, L.J.; Molkentin, J.D. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2). J. Cell Biol. 2002, 156, 905–919. [Google Scholar] [CrossRef]
- Yan, L.; Huang, H.; Tang, Q.-Z.; Zhu, L.-H.; Wang, L.; Liu, C.; Bian, Z.-Y.; Li, H. Breviscapine protects against cardiac hypertrophy through blocking PKC-α-dependent signaling. J. Cell. Biochem. 2010, 109, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.L.; Lam, C.S.P.; Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac endothelium–myocyte interaction: Clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur. Heart J. 2015, 36, 2050–2060. [Google Scholar] [CrossRef] [Green Version]
- Bry, M.; Kivelä, R.; Holopainen, T.; Anisimov, A.; Tammela, T.; Soronen, J.; Silvola, J.; Saraste, A.; Jeltsch, M.; Korpisalo, P.; et al. Vascular Endothelial Growth Factor-B Acts as a Coronary Growth Factor in Transgenic Rats Without Inducing Angiogenesis, Vascular Leak, or Inflammation. Circulation 2010, 122, 1725–1733. [Google Scholar] [CrossRef]
- Földes, G.; Suo, M.; Szokodi, I.; Lakó-Futó, Z.; deChâtel, R.; Vuolteenaho, O.; Huttunen, P.; Ruskoaho, H.; Tóth, M. Factors Derived from Adrenals Are Required for Activation of Cardiac Gene Expression in Angiotensin II-Induced Hypertension. Endocrinology 2001, 142, 4256–4263. [Google Scholar] [CrossRef]
- Joseph, J.; Claggett, B.C.; Anand, I.S.; Fleg, J.L.; Huynh, T.; Desai, A.S.; Solomon, S.D.; O’Meara, E.; Mckinlay, S.; Pitt, B.; et al. QRS Duration Is a Predictor of Adverse Outcomes in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 477–486. [Google Scholar] [CrossRef]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- van Berlo, J.H.; Maillet, M.; Molkentin, J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Investig. 2013, 123, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirronen, A.; Downes, N.L.; Huusko, J.; Laakkonen, J.P.; Tuomainen, T.; Tavi, P.; Hedman, M.; Ylä-Herttuala, S. The Ablation of VEGFR-1 Signaling Promotes Pressure Overload-Induced Cardiac Dysfunction and Sudden Death. Biomolecules 2021, 11, 452. https://doi.org/10.3390/biom11030452
Tirronen A, Downes NL, Huusko J, Laakkonen JP, Tuomainen T, Tavi P, Hedman M, Ylä-Herttuala S. The Ablation of VEGFR-1 Signaling Promotes Pressure Overload-Induced Cardiac Dysfunction and Sudden Death. Biomolecules. 2021; 11(3):452. https://doi.org/10.3390/biom11030452
Chicago/Turabian StyleTirronen, Annakaisa, Nicholas L. Downes, Jenni Huusko, Johanna P. Laakkonen, Tomi Tuomainen, Pasi Tavi, Marja Hedman, and Seppo Ylä-Herttuala. 2021. "The Ablation of VEGFR-1 Signaling Promotes Pressure Overload-Induced Cardiac Dysfunction and Sudden Death" Biomolecules 11, no. 3: 452. https://doi.org/10.3390/biom11030452
APA StyleTirronen, A., Downes, N. L., Huusko, J., Laakkonen, J. P., Tuomainen, T., Tavi, P., Hedman, M., & Ylä-Herttuala, S. (2021). The Ablation of VEGFR-1 Signaling Promotes Pressure Overload-Induced Cardiac Dysfunction and Sudden Death. Biomolecules, 11(3), 452. https://doi.org/10.3390/biom11030452