
Celecoxib Analogues for Cancer Treatment: An Update on OSU-03012 and 2,5-Dimethyl-Celecoxib



Abstract
:1. Introduction
2. From Adverse Side Effects of COX-2 Inhibitors toward Non-Active Celecoxib Analogs
3. Anti-Tumor Properties of OSU-03012 and DMC
3.1. Effects of Non-Active COX-2 Inhibitors Analogues on Cell Cycle Progression
3.1.1. 2,5-Dimethyl Celecoxib (DMC)
3.1.2. OSU-03012 (OSU)
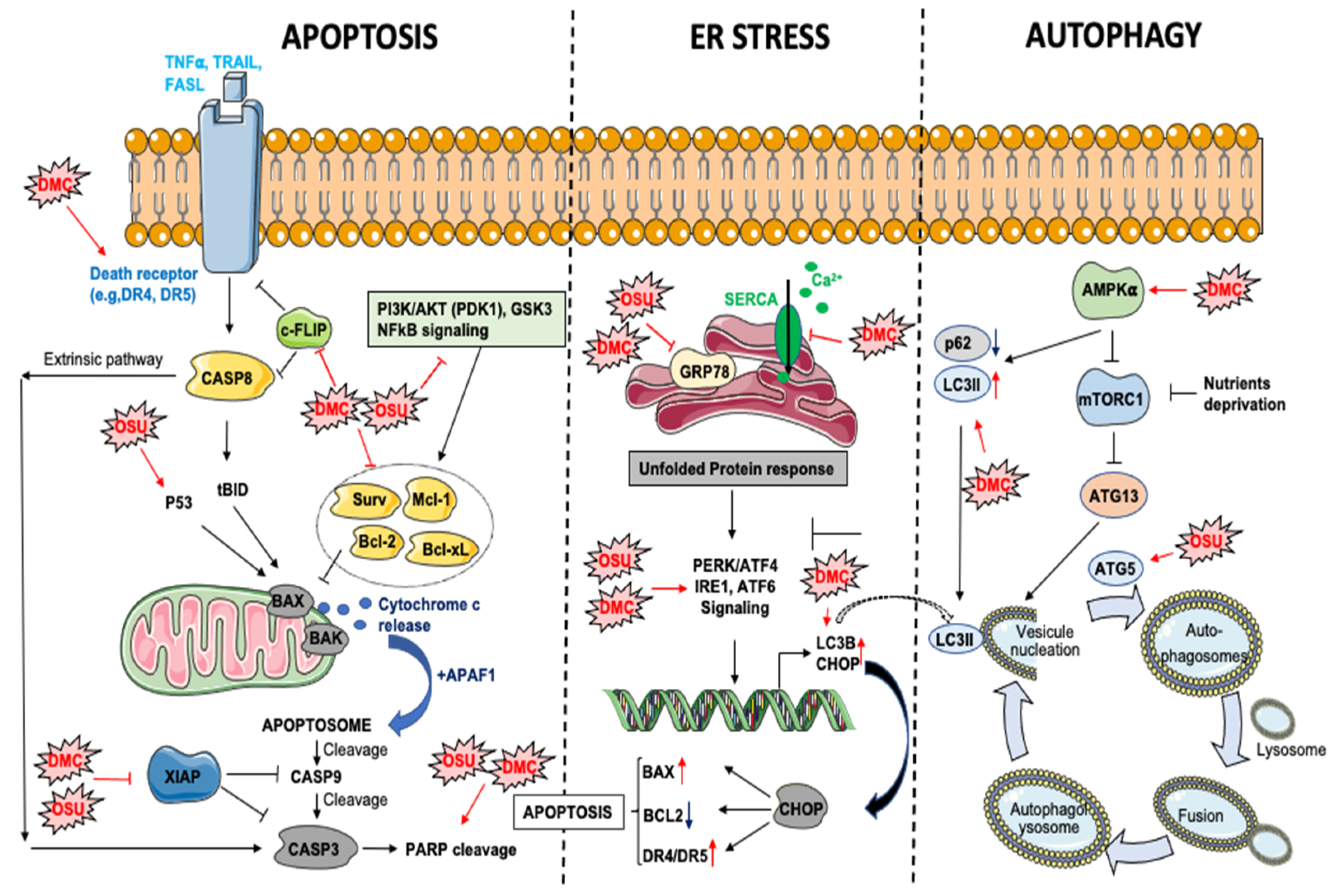
3.2. Effects of OSU and DMC on Apoptosis
3.2.1. Intrinsic Apoptosis
3.2.2. Extrinsic Apoptosis
3.2.3. DMC and OSU-03012 Are ER Stress Aggravators
3.3. DMC and OSU-03012 and Autophagy
3.4. Effects of OSU-03012 and DMC on Cancer Cells Migration/Invasion
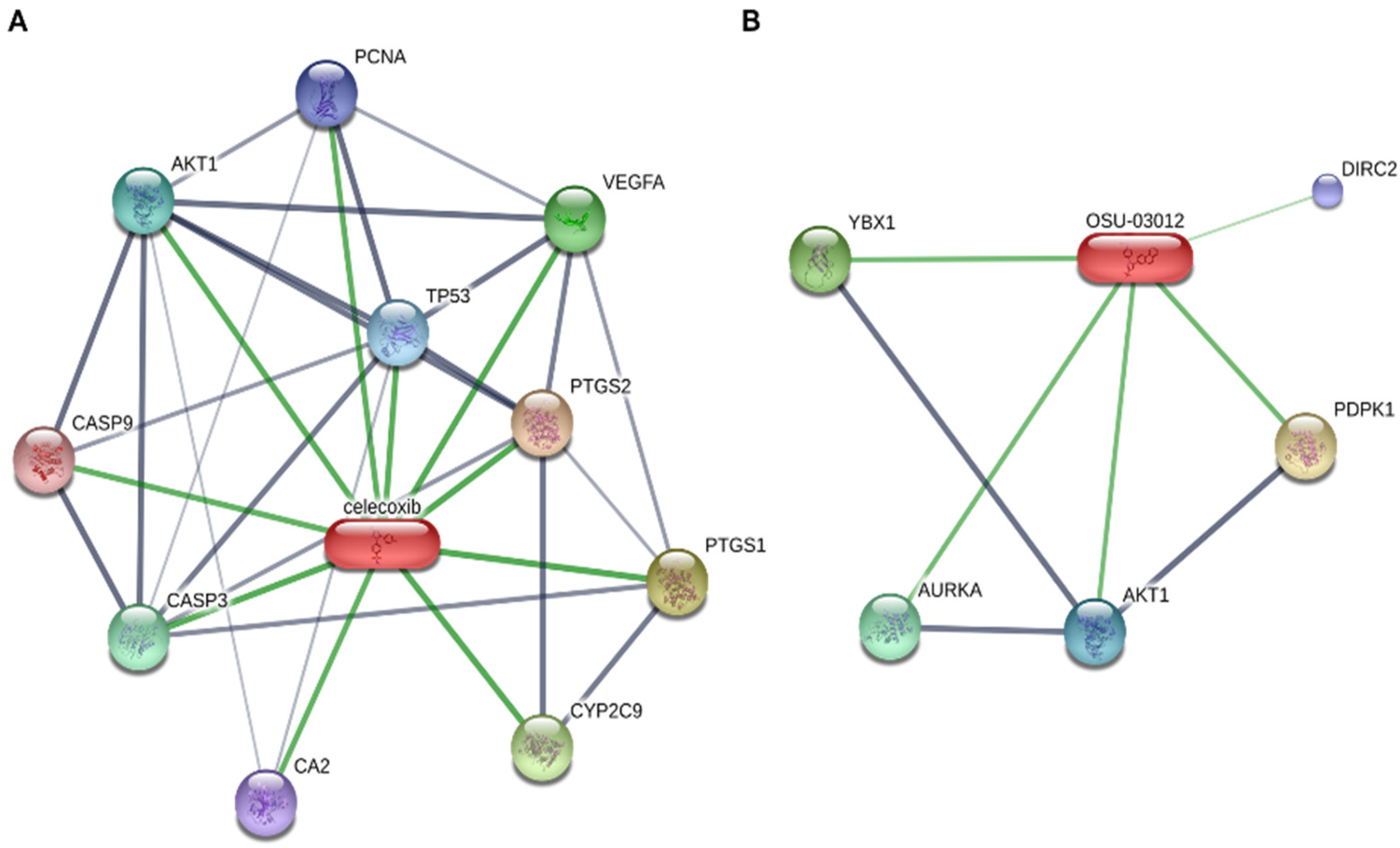
3.5. Other Potential Mechanisms?
4. Non-Active COX-2 Inhibitors in Combination with Other Therapeutic Approaches
4.1. DMC and OSU-03012 in Combination with Chemotherapy
4.2. DMC and OSU-03012 in Combination with Radiotherapy
4.3. DMC and OSU-03012 in Combination with Photodynamic Therapy
4.4. Other Potential Combinations

{kind=link}
{kind=link}
{kind=link}
| Combinations | Cancer Type | Models Used | Mechanisms Behind a Better Response | References |
|---|---|---|---|---|
| OSU-03012/ Cisplatin | Ovarian Cancer | Cisplatin resistant A2780 cells | Increased apoptosis | [80] |
| DMC/ Perillyl Alcohol | Glioma | Temozolomide-sensitive and -resistant glioma cells | ER stress induction | [99] |
| OSU-03012/ Radiotherapy | Glioblastoma | Glioblastoma cancer cells | ER stress induction | [82] |
| CRC | CRC cells: HCT-116 | ER stress induction | [83] | |
| DMC/PDT (Photofrin) | Breast Cancer | BA cells | ER stress induction | [100] |
| Survivin downregulation | ||||
| DMC/ Chloroquin/ Nelfinavir | Breast Cancer | MDA-MB-231, MDA-MB-468 | PARP, Caspase 7 and -3 cleavage | [30] |
| Aggravation of ER stress | ||||
| Autophagy induction | ||||
| OSU-03012/ Imatinib mesylate | CML | (Bcr)-Abl mutant cell lines: Ba/F3p210(E255K) Ba/F3p210(T315I) | PI3K/AKT pathway inhibition | [87] |
| Myeloma | TIB-196 myeloma cells | Phospho-STAT3 downregulation | [88] | |
| Increased phospho-AMPK (Thr172) | ||||
| DMC/ Imatinib mesylate | CRC | HT-29 cells | Undetermined | [89] |
| OSU-03012/ Lapatinib | Breast Cancer | MDA-MB-231 cell line | Increased Eif2α phosphorylation Nck1downregulation | [91] |
| Glioblastoma | GBM5,6, 12, 14 cells | Inhibition of multiple ERBB receptors | [42] | |
| OSU-03012/ Trastuzumab | Breast Cancer | HER2-expressing breast cancer cells | Impairment of PDK-1/AKT signaling | [92] |
| DMC/ Trastuzumab | HCC | HBV positive hepatoma | Inhibition of PD-1/PD-L1 pathway | [79]. |
| DMC/ABT-737 | Gastric Cancer | AGS and HGC-27 cells | ER stress induction | [54] |
| OSU/Sildenafil | Glioblastoma | GBM5,6, 12, 14 cells | Death receptor signaling ER stress response | [61] |
5. Are Non-Active COX-2 Inhibitors Devoid of Adverse Side Effects?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Stein-Werblowsky, R. Prostaglandin and Cancer. Oncology 1974, 30, 169–176. [Google Scholar] [CrossRef]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a Double-Edge Sword for Cancer and Other Age-Related Diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Li, N.; Fu, J.; Sun, Y.; Li, Y.; Qu, R.; Jin, X.; Li, D. Upregulation of cyclooxygenase-2 is associated with activation of the alternative nuclear factor kappa B signaling pathway in colonic adenocarcinoma. Am. J. Transl. Res. 2015, 7, 1612–1620. [Google Scholar] [PubMed]
- Liu, Y.; Li, H.; Zhao, C.; Jia, H. MicroRNA-101 inhibits angiogenesis via COX-2 in endometrial carcinoma. Mol. Cell. Biochem. 2018, 448, 61–69. [Google Scholar] [CrossRef]
- Mahboubi-Rabbani, M.I.; Zarghi, A. Selective COX-2 inhibitors as anticancer agents: A patent review (2014–2018). Expert Opin. Ther. Pat. 2019, 29, 407–427. [Google Scholar] [CrossRef]
- Tolloczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Lynch, P.M. Chemoprevention of familial adenomatous polyposis. Fam. Cancer 2016, 15, 467–475. [Google Scholar] [CrossRef]
- Grösch, S.; Tegeder, I.; Niederberger, E.; Bräutigam, L.; Geisslinger, G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J. 2001, 15, 1–22. [Google Scholar] [CrossRef]
- Gallouet, A.-S.; Travert, M.; Bresson-Bepoldin, L.; Guilloton, F.; Pangault, C.; Caulet-Maugendre, S.; Lamy, T.; Tarte, K.; Guillaudeux, T. COX-2–Independent Effects of Celecoxib Sensitize Lymphoma B Cells to TRAIL-Mediated Apoptosis. Clin. Cancer Res. 2014, 20, 2663–2673. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Leng, J.; Demetris, A.J.; Wu, T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: Evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004, 64, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Romagnolo, D.F.; Papoutsis, A.J.; Selmin, O. Nutritional targeting of cyclooxygenase-2 for colon cancer prevention. Inflamm. Allergy Drug Targets 2010, 9, 181–191. [Google Scholar] [CrossRef]
- Fitzpatrick, F.A. Cyclooxygenase Enzymes: Regulation and Function. Curr. Pharm. Des. 2004, 10, 577–588. [Google Scholar] [CrossRef]
- Nørregaard, R.; Kwon, T.-H.; Frøkiær, J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res. Clin. Pract. 2015, 34, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Peskar, B.M.; Maricic, N.; Gretzer, B.; Schuligoi, R.; Schmassmann, A. Role of cyclooxygenase-2 in gastric mucosal defense. Life Sci. 2001, 69, 2993–3003. [Google Scholar] [CrossRef]
- Sharma, J.N.; Jawad, N.M. Adverse Effects of COX-2 Inhibitors. Sci. World J. 2005, 5, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Qiu, H.; Haeggström, J.Z.; Sakata, K. Leukotriene B4is an indirectly acting vasoconstrictor in guinea pig aorta via an inducible type of BLT receptor. Am. J. Physiol. Circ. Physiol. 2004, 287, H419–H424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montuschi, P.; Macagno, F.; Parente, P.; Valente, S.; Lauriola, L.; Ciappi, G.; A Kharitonov, S.; Barnes, P.J.; Ciabattoni, G. Effects of cyclo-oxygenase inhibition on exhaled eicosanoids in patients with COPD. Thorax 2005, 60, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnier, M. The safety of rofecoxib. Expert Opin. Drug Saf. 2005, 4, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Jüni, P.; Nartey, L.; Reichenbach, S.; Sterchi, R.; A Dieppe, P.; Egger, M. Risk of cardiovascular events and rofecoxib: Cumulative meta-analysis. Lancet 2004, 364, 2021–2029. [Google Scholar] [CrossRef]
- Yang, C.F.; Gopula, B.; Liang, J.J.; Li, J.K.; Chen, S.Y.; Lee, Y.L.; Chen, C.S.; Lin, Y.L. Novel AR-12 derivatives, P12-23 and P12-34, inhibit flavivirus replication by blocking host de novo pyrimidine biosynthesis. Emerg. Microbes Infect. 2018, 7, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.-W.; Zhu, Z.; Chu, H.; Yuan, S.; Chik, K.K.-H.; Chan, C.C.-S.; Poon, V.K.-M.; Yip, C.C.-Y.; Zhang, X.; Tsang, J.O.-L.; et al. The celecoxib derivative kinase inhibitor AR-12 (OSU-03012) inhibits Zika virus via down-regulation of the PI3K/Akt pathway and protects Zika virus-infected A129 mice: A host-targeting treatment strategy. Antivir. Res. 2018, 160, 38–47. [Google Scholar] [CrossRef]
- Park, J.-G.; Ávila-Pérez, G.; Nogales, A.; Blanco-Lobo, P.; de la Torre, J.C.; Martínez-Sobrido, L. Identification and Characterization of Novel Compounds with Broad-Spectrum Antiviral Activity against Influenza A and B Viruses. J. Virol. 2020, 94, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, J.O.; Roberts, R.A.; Kim, J.; Poklepovic, A.; Roberts, J.L.; Booth, L.; Dent, P. AR12 (OSU-03012) suppresses GRP78 expression and inhibits SARS-CoV-2 replication. Biochem. Pharmacol. 2020, 182, 114227. [Google Scholar] [CrossRef]
- Barker, W.T.; Nemeth, A.M.; Brackett, S.M.; Basak, A.K.; Chandler, C.; Jania, L.A.; Zuercher, W.J.; Melander, R.J.; Koller, B.H.; Ernst, R.; et al. Repurposing Eukaryotic Kinase Inhibitors as Colistin Adjuvants in Gram-Negative Bacteria. ACS Infect. Dis. 2019, 5, 1764–1771. [Google Scholar] [CrossRef]
- Egashira, I.; Takahashi-Yanaga, F.; Nishida, R.; Arioka, M.; Igawa, K.; Tomooka, K.; Nakatsu, Y.; Tsuzuki, T.; Nakabeppu, Y.; Kitazono, T.; et al. Celecoxib and 2,5-dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β-catenin signaling pathway. Cancer Sci. 2016, 108, 108–115. [Google Scholar] [CrossRef]
- E Kucab, J.; Lee, C.; Chen, C.-S.; Zhu, J.; Gilks, C.B.; Cheang, M.C.U.; Huntsman, D.; Yorida, E.; Emerman, J.; Pollak, M.; et al. Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res. 2005, 7, R796. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Sharma, N.; Golden, E.B.; Cho, H.; Agarwal, P.; Gaffney, K.; Petasis, N.; Chen, T.C.; Hofman, F.M.; Louie, S.G.; et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012, 325, 63–71. [Google Scholar] [CrossRef]
- Johnson, A.J.; Smith, L.L.; Zhu, J.; Heerema, N.A.; Jefferson, S.; Mone, A.; Grever, M.; Chen, C.-S.; Byrd, J.C. A novel celecoxib derivative, OSU03012, induces cytotoxicity in primary CLL cells and transformed B-cell lymphoma cell line via a caspase- and Bcl-2–independent mechanism. Blood 2005, 105, 2504–2509. [Google Scholar] [CrossRef]
- Li, J.; Zhu, J.; Melvin, W.S.; Bekaii-Saab, T.S.; Chen, C.-S.; Muscarella, P. A Structurally Optimized Celecoxib Derivative Inhibits Human Pancreatic Cancer Cell Growth. J. Gastrointest. Surg. 2006, 10, 207–214. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, J.W.; Tseng, P.H.; Yang, Y.T.; Fowble, J.; Shiau, C.W.; Shaw, Y.J.; Kulp, S.K.; Chen, C.S. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004, 64, 4309–4318. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Moon, H.; Lee, Y.; Kang, C.; Kim, S. Potentiation of TRAIL-induced cell death by nonsteroidal anti-inflammatory drug in human hepatocellular carcinoma cells through the ER stress-dependent autophagy pathway. Oncol. Rep. 2020, 44, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yeh, P.Y.; Lu, Y.-S.; Hsu, C.-H.; Chen, K.-F.; Lee, W.-C.; Feng, W.-C.; Chen, C.-S.; Kuo, M.-L.; Cheng, A.-L. OSU-03012, a Novel Celecoxib Derivative, Induces Reactive Oxygen Species–Related Autophagy in Hepatocellular Carcinoma. Cancer Res. 2008, 68, 9348–9357. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-Z.; Liu, X.; Yue, P.; Schönthal, A.H.; Khuri, F.R.; Sun, S.-Y. CCAAT/Enhancer Binding Protein Homologous Protein-Dependent Death Receptor 5 Induction and Ubiquitin/Proteasome-Mediated Cellular FLICE-Inhibitory Protein Down-Regulation Contribute to Enhancement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis by Dimethyl-Celecoxib in Human Non–Small-Cell Lung Cancer Cells. Mol. Pharmacol. 2007, 72, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Roosmalen, I.A.M.V.; Reis, C.R.; Setroikromo, R.; Yuvaraj, S.; Joseph, J.V.; Tepper, P.G.; A E Kruyt, F.; Quax, W.J. The ER stress inducer DMC enhances TRAIL-induced apoptosis in glioblastoma. SpringerPlus 2014, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Nyalali, A.M.K.; Hou, Y.; Xu, Y.; Zhou, J.; Zhao, W.; Huang, B.; Li, F. 2,5-Dimethyl Celecoxib Inhibits Proliferation and Cell Cycle and Induces Apoptosis in Glioblastoma by Suppressing CIP2A/PP2A/Akt Signaling Axis. J. Mol. Neurosci. 2021, 1–11. [Google Scholar] [CrossRef]
- Booth, L.; Cazanave, S.C.; Hamed, H.A.; Yacoub, A.; Ogretmen, B.; Chen, C.-S.; Grant, S.; Dent, P. OSU-03012 suppresses GRP78/BiP expression that causes PERK-dependent increases in tumor cell killing. Cancer Biol. Ther. 2012, 13, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, R.; Devi, A.; Mishra, S. Molecular docking and molecular dynamics studies reveal structural basis of inhibition and selectivity of inhibitors EGCG and OSU-03012 toward glucose regulated protein-78 (GRP78) overexpressed in glioblastoma. J. Mol. Model. 2015, 21, 272. [Google Scholar] [CrossRef] [PubMed]
- Hamed, H.A.; Yacoub, A.; Park, M.A.; Eulitt, P.J.; Sarkar, D.; Dimitriev, I.P.; Chen, C.-S.; Grant, S.; Curiel, D.T.; Fisher, P.B.; et al. OSU-03012 enhances Ad.7-induced GBM cell killing via ER stress and autophagy and by decreasing expression of mitochondrial protective proteins. Cancer Biol. Ther. 2010, 9, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.; Cruickshanks, N.; Ridder, T.; Chen, C.-S.; Grant, S.; Dent, P. OSU-03012 interacts with lapatinib to kill brain cancer cells. Cancer Biol. Ther. 2012, 13, 1501–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Qin, C.-K.; Lv, W.; Zhao, Q. OSU-03012, a non-cox inhibiting celecoxib derivative, induces apoptosis of human esophageal carcinoma cells through a p53/Bax/cytochrome c/caspase-9-dependent pathway. Anti-Cancer Drugs 2013, 24, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Kardosh, A.; Wang, W.; Uddin, J.; Petasis, N.A.; Hofman, F.M.; Chen, T.C.; Schonthal, A.H. Dimethyl-Celecoxib (DMC), a derivative of celecoxib that lacks cyclooxygenase-2-Inhibitory function, potently mimics the anti-tumor effects of celecoxib on burkitt’s lymphoma in vitro and in vivo. Cancer Biol. Ther. 2005, 4, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolewski, C.; Rhim, J.; Legrand, N.; Muller, F.; Cerella, C.; Mack, F.; Chateauvieux, S.; Kim, J.-G.; Yoon, A.-Y.; Kim, K.-W.; et al. 2,5-Dimethyl-Celecoxib Inhibits Cell Cycle Progression and Induces Apoptosis in Human Leukemia Cells. J. Pharmacol. Exp. Ther. 2015, 355, 308–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porchia, L.M.; Guerra, M.; Wang, Y.-C.; Zhang, Y.; Espinosa, A.V.; Shinohara, M.; Kulp, S.K.; Kirschner, L.S.; Saji, M.; Chen, C.-S.; et al. 2-Amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phenyl} Acetamide (OSU-03012), a Celecoxib Derivative, Directly Targets p21-Activated Kinase. Mol. Pharmacol. 2007, 72, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Vader, G.; Kauw, J.J.W.; Medema, R.H.; A Lens, S.M. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006, 7, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, H.; Qiu, Q.; Zhang, Z.; Gu, Y.; He, Z. TCRP1 promotes NIH/3T3 cell transformation by over-activating PDK1 and AKT1. Oncogenesis 2017, 6, e323. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Jin, L.; Lei, X.; Tong, J.; Wang, R. MicroRNA-363-3p inhibits cell proliferation and induces apoptosis in retinoblastoma cells via the Akt/mTOR signaling pathway by targeting PIK3CA. Oncol. Rep. 2020, 43, 1365–1374. [Google Scholar] [CrossRef]
- Silva, A.; Wang, J.; Lomahan, S.; Tran, T.-A.; Grenlin, L.; Suganami, A.; Tamura, Y.; Ikegaki, N. Aurora kinase A is a possible target of OSU-03012 to destabilize MYC family proteins. Oncol. Rep. 2014, 32, 901–905. [Google Scholar] [CrossRef] [Green Version]
- Abt, E.R.; Rosser, E.W.; Durst, M.A.; Lok, V.; Poddar, S.; Le, T.M.; Cho, A.; Kim, W.; Wei, L.; Song, J.; et al. Metabolic Modifier Screen Reveals Secondary Targets of Protein Kinase Inhibitors within Nucleotide Metabolism. Cell Chem. Biol. 2020, 27, 197–205.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, M.; Zhang, Z.; Luo, Z.; Liu, F.; Liu, J. Celecoxib derivative OSU-03012 inhibits the proliferation and activation of hepatic stellate cells by inducing cell senescence. Mol. Med. Rep. 2015, 11, 3021–3026. [Google Scholar] [CrossRef] [PubMed]
- Nikanfar, S.; Atari-Hajipirloo, S.; Kheradmand, F.; Rashedi, J.; Heydari, A. Cytotoxic effect of 2, 5-dimethyl-celecoxib as a structural analog of celecoxib on human colorectal cancer (HT-29) cell line. Cell Mol. Biol. 2018, 64, 8–13. [Google Scholar] [CrossRef]
- Zhang, B.; Yan, Y.; Li, Y.; Zhang, D.; Zeng, J.; Wang, L.; Wang, M.; Lin, N. Dimethyl celecoxib sensitizes gastric cancer cells to ABT-737 via AIF nuclear translocation. J. Cell Mol. Med. 2016, 20, 2148–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Suvannasankha, A.; Crean, C.D.; White, V.L.; Johnson, A.; Chen, C.-S.; Farag, S.S. OSU-03012, a Novel Celecoxib Derivative, Is Cytotoxic to Myeloma Cells and Acts through Multiple Mechanisms. Clin. Cancer Res. 2007, 13, 4750–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virrey, J.J.; Liu, Z.; Cho, H.-Y.; Kardosh, A.; Golden, E.B.; Louie, S.G.; Gaffney, K.; Petasis, N.; Schönthal, A.H.; Chen, T.C.; et al. Antiangiogenic Activities of 2,5-Dimethyl-Celecoxib on the Tumor Vasculature. Mol. Cancer Ther. 2010, 9, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Tavallai, M.; Nourbakhsh, A.; Chuckalovcak, J.; Carter, J.; Poklepovic, A.; Dent, P. OSU-03012 and Viagra Treatment Inhibits the Activity of Multiple Chaperone Proteins and Disrupts the Blood–Brain Barrier: Implications for Anti-Cancer Therapies. J. Cell. Physiol. 2015, 230, 1982–1998. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Grant, S.; Poklepovic, A.; Dent, P. Regulation of OSU-03012 Toxicity by ER Stress Proteins and ER Stress–Inducing Drugs. Mol. Cancer Ther. 2014, 13, 2384–2398. [Google Scholar] [CrossRef] [Green Version]
- Pyrko, P.; Kardosh, A.; Liu, Y.-T.; Soriano, N.; Xiong, W.; Chow, R.H.; Uddin, J.; Petasis, N.; Mircheff, A.K.; Farley, R.A.; et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Mol. Cancer Ther. 2007, 6, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138. [Google Scholar] [CrossRef]
- Corti, O.; Blomgren, K.; Poletti, A.; Beart, P.M. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J. Neurochem. 2020, 154, 354–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, 219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Wang, Y.; Xiang, Y.; Tanaka, K.E.; Gaarde, W.A.; Czaja, M.J. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology 2009, 49, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Singh, R.; Massey, A.C.; Kane, S.S.; Kaushik, S.; Grant, T.; Xiang, Y.; Cuervo, A.M.; Czaja, M.J. Loss of Macroautophagy Promotes or Prevents Fibroblast Apoptosis Depending on the Death Stimulus. J. Biol. Chem. 2008, 283, 4766–4777. [Google Scholar] [CrossRef] [Green Version]
- Backhus, L.M.; Petasis, N.; Uddin, J.; Schönthal, A.H.; Bart, R.D.; Lin, Y.; Starnes, V.A.; Bremner, R.M. Dimethyl celecoxib as a novel non–cyclooxygenase 2 therapy in the treatment of non–small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2005, 130, 1406–1412. [Google Scholar] [CrossRef] [Green Version]
- Alcántara-Hernández, R.; Hernández-Méndez, A.; García-Sáinz, J.A. The phosphoinositide-dependent protein kinase 1 inhibitor, UCN-01, induces fragmentation: Possible role of metalloproteinases. Eur. J. Pharmacol. 2014, 740, 88–96. [Google Scholar] [CrossRef]
- Ma, Y.; Mccarty, S.K.; Kapuriya, N.P.; Brendel, V.J.; Wang, C.; Zhang, X.; Jarjoura, D.; Saji, M.; Chen, C.-S.; Ringel, M.D. Development of p21 Activated Kinase-Targeted Multikinase Inhibitors That Inhibit Thyroid Cancer Cell Migration. J. Clin. Endocrinol. Metab. 2013, 98, E1314–E1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönthal, A.H. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: Implications for glioma therapy. Neurosurg. Focus 2006, 20, E21. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.G.T.; Marschner, G.; Canny, G.; Klocke, S.; Hunold, P.; Köster, F.; Ahrens, T.; Rody, A.; Hornung, D. Disrupting Y-Box-Binding Protein 1 Function Using OSU-03012 Prevents Endometriosis Progression in In Vitro and In Vivo Models. Reprod. Sci. 2016, 24, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, H.; Wu, L.; Huang, S. YBX1 promotes tumor growth by elevating glycolysis in human bladder cancer. Oncotarget 2017, 8, 65946–65956. [Google Scholar] [CrossRef] [Green Version]
- Kuwano, M.; Shibata, T.; Watari, K.; Ono, M. Oncogenic Y-box binding protein-1 as an effective therapeutic target in drug-resistant cancer. Cancer Sci. 2019, 110, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Savalas, L.R.T.; Gasnier, B.; Damme, M.; Lübke, T.; Wrocklage, C.; Debacker, C.; Jezegou, A.; Reinheckel, T.; Hasilik, A.; Saftig, P.; et al. Disrupted in renal carcinoma 2 (DIRC2), a novel transporter of the lysosomal membrane, is proteolytically processed by cathepsin L. Biochem. J. 2011, 439, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chen, Y.; Peng, L.; Wang, X.; Tang, N. 2,5-dimethylcelecoxib improves immune microenvironment of hepatocellular carcinoma by promoting ubiquitination of HBx-induced PD-L1. J. Immunother. Cancer 2020, 8, e001377. [Google Scholar] [CrossRef] [PubMed]
- Sima, N.; Sun, W.; Gorshkov, K.; Shen, M.; Huang, W.; Zhu, W.; Xie, X.; Zheng, W.; Cheng, X. Small Molecules Identified from a Quantitative Drug Combinational Screen Resensitize Cisplatin’s Response in Drug-Resistant Ovarian Cancer Cells. Transl. Oncol. 2018, 11, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Her, S.; Jaffray, D. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. OSU-03012 in the Treatment of Glioblastoma. Mol. Pharmacol. 2006, 70, 437–439. [Google Scholar] [CrossRef]
- Caron, R.W.; Yacoub, A.; Li, M.; Zhu, X.; Mitchell, C.; Hong, Y.; Hawkins, W.; Sasazuki, T.; Shirasawa, S.; Kozikowski, A.P.; et al. Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol. Cancer Ther. 2005, 4, 257–270. [Google Scholar] [PubMed]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Ferrario, A.; Fisher, A.M.; Rucker, N.; Gomer, C.J. Celecoxib and NS-398 Enhance Photodynamic Therapy by Increasing In vitro Apoptosis and Decreasing In vivo Inflammatory and Angiogenic Factors. Cancer Res. 2005, 65, 9473–9478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, A.; Lim, S.; Xu, F.; Luna, M.; Gaffney, K.J.; Petasis, N.A.; Schönthal, A.H.; Gomer, C.J. Enhancement of photodynamic therapy by 2,5-dimethyl celecoxib, a non-cyclooxygenase-2 inhibitor analog of celecoxib. Cancer Lett. 2011, 304, 33–40. [Google Scholar] [CrossRef]
- Tseng, P.-H.; Lin, H.-P.; Zhu, J.; Chen, K.-F.; Hade, E.M.; Young, D.C.; Byrd, J.C.; Grever, M.; Johnson, K.; Druker, B.J.; et al. Synergistic interactions between imatinib mesylate and the novel phosphoinositide-dependent kinase-1 inhibitor OSU-03012 in overcoming imatinib mesylate resistance. Blood 2005, 105, 4021–4027. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.-Y.; Weng, J.-R.; Tsai, C.-H.; Sargeant, A.; Lin, C.-W.; Chiu, C.-F. OSU-03012 sensitizes TIB-196 myeloma cells to imatinib mesylate via AMP-activated protein kinase and STAT3 pathways. Leuk. Res. 2010, 34, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Atari-Hajipirloo, S.; Nikanfar, S.; Heydari, A.; Kheradmand, F. Imatinib and its combination with 2,5-dimethyl-celecoxibinduces apoptosis of human HT-29 colorectal cancer cells. Res. Pharm. Sci. 2017, 12, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Mughal, T. Principal long-term adverse effects of imatinib in patients with chronic myeloid leukemia in chronic phase. Biol. Targets Ther. 2010, 4, 315–323. [Google Scholar] [CrossRef] [Green Version]
- West, N.W.; Garcia-Vargas, A.; E Chalfant, C.; A Park, M. OSU-03012 sensitizes breast cancers to lapatinib-induced cell killing: A role for Nck1 but not Nck2. BMC Cancer 2013, 13, 1–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, P.-H.; Wang, Y.-C.; Weng, S.-C.; Weng, J.-R.; Chen, C.-S.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.-Y.; Dunn, S.E.; Pollak, M. Overcoming Trastuzumab Resistance in HER2-Overexpressing Breast Cancer Cells by Using a Novel Celecoxib-Derived Phosphoinositide-Dependent Kinase-1 Inhibitor. Mol. Pharmacol. 2006, 70, 1534–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpel-Massler, G.; Ishida, C.T.; Zhang, Y.; Halatsch, M.-E.; Westhoff, M.-A.; Siegelin, M.D. Targeting intrinsic apoptosis and other forms of cell death by BH3-mimetics in glioblastoma. Expert Opin. Drug Discov. 2017, 12, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharmacol. 2017, 136, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Lieber, J.; Armeanu-Ebinger, S.; Fuchs, J. The Role of BH3-Mimetic Drugs in the Treatment of Pediatric Hepatoblastoma. Int. J. Mol. Sci. 2015, 16, 4190–4208. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, S.; Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Mcl-1 Phosphorylation Defines ABT-737 Resistance That Can Be Overcome by Increased NOXA Expression in Leukemic B cells. Cancer Res. 2012, 72, 3069–3079. [Google Scholar] [CrossRef] [Green Version]
- Anstee, N.; Bilardi, R.A.; Ng, A.P.; Xu, Z.; Robati, M.; Vandenberg, C.J.; Cory, S. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 AML in mice. Cell Death Differ. 2019, 26, 1316–1331. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Whittle, J.R.; Vaillant, F.; Serrano, A.; Gong, J.-N.; Giner, G.; Maragno, A.L.; Chanrion, M.; Schneider, E.; Pal, B.; et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 2017, 9, eaam7049. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-Y.; Wang, W.; Jhaveri, N.; Torres, S.; Tseng, J.; Leong, M.N.; Lee, D.J.; Goldkorn, A.; Xu, T.; Petasis, N.; et al. Perillyl Alcohol for the Treatment of Temozolomide-Resistant Gliomas. Mol. Cancer Ther. 2012, 11, 2462–2472. [Google Scholar] [CrossRef] [Green Version]
- Dowidar, A.; Abdel-Monem, M.H. Effect of chemical pollutants on bacterial counts in El-Temsah Lake area, Ismailia, Egypt. J. Egypt. Public Health Assoc. 1990, 65, 305–318. [Google Scholar]
- Kulp, S.K.; Yang, Y.T.; Hung, C.C.; Chen, K.F.; Lai, J.P.; Tseng, P.H.; Fowble, J.W.; Ward, P.J.; Chen, C.S. 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res. 2004, 64, 1444–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, K.V.; Borteh, H.M.; Rajaram, M.V.; Peine, K.J.; Curry, H.; Collier, M.A.; Homsy, M.L.; Bachelder, E.M.; Gunn, J.S.; Schlesinger, L.S.; et al. Acetalated dextran encapsulated AR-12 as a host-directed therapy to control Salmonella infection. Int. J. Pharm. 2014, 477, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Collier, M.A.; Peine, K.J.; Gautam, S.; Oghumu, S.; Varikuti, S.; Borteh, H.; Papenfuss, T.L.; Sataoskar, A.R.; Bachelder, E.M.; Ainslie, K.M. Host-mediated Leishmania donovani treatment using AR-12 encapsulated in acetalated dextran microparticles. Int. J. Pharm. 2016, 499, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Lian, T.; Fan, X.; Song, C.; Gaur, U.; Mao, X.; Yang, D.; Piper, M.D.W.; Yang, M. 2,5-Dimethyl-Celecoxib Extends Drosophila Life Span via a Mechanism That Requires Insulin and Target of Rapamycin Signaling. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1334–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckmann, K.; Rorsch, F.; Steri, R.; Schubert-Zsilavecz, M.; Geisslinger, G.; Grosch, S. Dimethylcelecoxib inhibits mPGES-1 promoter activity by influencing EGR1 and NF-kappaB. Biochem. Pharmacol. 2010, 80, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Wobst, I.; Schiffmann, S.; Birod, K.; Maier, T.J.; Schmidt, R.; Angioni, C.; Geisslinger, G.; Grosch, S. Dimethylcelecoxib inhibits prostaglandin E2 production. Biochem. Pharmacol. 2008, 76, 62–69. [Google Scholar] [CrossRef]
- Yamamoto, S.; Iyoda, T.; Kita, S.; Yamada, T.; Iwamoto, T. OSU-03012, a novel celecoxib derivative, induces cell swelling and shortens action potential duration in mouse ventricular cells. Biomed. Res. 2010, 31, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Morishige, S.; Takahashi-Yanaga, F.; Ishikane, S.; Arioka, M.; Igawa, K.; Kuroo, A.; Tomooka, K.; Shiose, A.; Sasaguri, T. 2,5-Dimethylcelecoxib prevents isoprenaline-induced cardiomyocyte hypertrophy and cardiac fibroblast activation by inhibiting Akt-mediated GSK-3 phosphorylation. Biochem. Pharmacol. 2019, 168, 82–90. [Google Scholar] [CrossRef]
- Fan, X.; Takahashi-Yanaga, F.; Morimoto, S.; Zhan, D.Y.; Igawa, K.; Tomooka, K.; Sasaguri, T. Celecoxib and 2,5-dimethyl-celecoxib prevent cardiac remodeling inhibiting Akt-mediated signal transduction in an inherited dilated cardiomyopathy mouse model. J. Pharmacol. Exp. Ther. 2011, 338, 2–11. [Google Scholar] [CrossRef]
- Fujita, A.; Takahashi-Yanaga, F.; Morimoto, S.; Yoshihara, T.; Arioka, M.; Igawa, K.; Tomooka, K.; Hoka, S.; Sasaguri, T. 2,5-Dimethylcelecoxib prevents pressure-induced left ventricular remodeling through GSK-3 activation. Hypertens. Res. 2017, 40, 130–139. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takahashi-Yanaga, F.; Arioka, M.; Igawa, K.; Tomooka, K.; Yamaura, K.; Sasaguri, T. Cardiac and renal protective effects of 2,5-dimethylcelecoxib in angiotensin II and high-salt-induced hypertension model mice. J. Hypertens. 2021, 39, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, K.A.; Donnelly, M.P.; Gee, T.W.; Ibrahim Aibo, M.A.; Byers, S.; Butcher, J.T. Induction of aortic valve calcification by celecoxib and its COX-2 independent derivatives is glucocorticoid-dependent. Cardiovasc. Pathol. 2020, 46, 107194. [Google Scholar] [CrossRef] [PubMed]



| Cancers | Molecule | Processes | Targets | Expression | Models | References |
|---|---|---|---|---|---|---|
| Colorectal Cancer | DMC | Induces Apoptosis | Survivin |  | HCT-116 cells | [28] |
| Inhibits cell cycle | Cyclin D1 | | ||||
| Breast Cancer | OSU | Induces Apoptosis | p-AKT | | MDA-MB-453, MCF-7, T47D, MDA-MB-231 and HBL100 | [29] |
| PARP cleavage |  | |||||
| DMC + Chloroquine and Nelfinavir | Induces Apoptosis | PARP cleavage | | MDA-MB-231, MDA-MB-468, Hs578t, T47D and MCF-7 | [30] | |
| Induces ER stress | GRP78, CHOP | | ||||
| CLL | OSU | Induces Intrinsic Apoptosis | Cleaved caspase 3/9 and PARP | | PBMCs from patients with CLL | [31] |
| Pancreatic cancer | OSU | Inhibits Migration/ Invasion | PDK1/p-AKT | | sPC-1, BxPC-3, Mia- PaCa 2, and PANC-1 | [32] |
| Prostate cancer | OSU | Inhibits Cell cycle Induces Apoptosis | PDK1/AKT signaling | | PC-3 cells | [33] |
| PDK1 expression | | |||||
| Hepatocellular carcinoma | DMC | Increases Autophagy | LC3II | | SNU-354, SNU-423, SNU-449 and SNU-475 cells | [34] |
| p-AMPK | | |||||
| p-mTOR | | |||||
| Induces ER stress | CHOP | | ||||
| Induces extrinsic Apoptosis | c-FLIP | | ||||
| DR5 | | |||||
| OSU | Induces Autophagy | ROS | | Huh7, Hep3B and HepG2 cells | [35] | |
| ATG5 expression | | |||||
| Inhibits Growth, Cell cycle | Undetermined | - | ||||
| Lung cancer | DMC | Induces apoptosis | CHOP | | Human Non-Small-Cell Lung Cancer cells | [36] |
| DR5 | | |||||
| c-FLIP | | |||||
| Glioblastoma | DMC | Induces Apoptosis | Survivin | | A172, U87, SNB75 and U251 cells | [37] |
| Cleaved caspase 3 and PARP | | |||||
| Inhibits Cell cycle Induces apoptosis | p21 | | LN229, U87MG, A172, and U251 cells | [38] | ||
| CIP2A/PP2A/AKT | | |||||
| Induces ER stress | GRP78 | | ||||
| CHOP | | |||||
| OSU | Induces ER stress | GRP78/Bip | | GBM12, GBM14 GBM6-luciferase cells (xenograft in mice) | [39] | |
| p-PERK | | |||||
| Increased Autophagy | LC3II | | U-373 MG and U-87 MG | [40] | ||
| Induces Apoptosis | MCL1 | | Primary GBM cells, GBM6, 12, 14 cells | [41] | ||
| BcLxL | | |||||
| PI3K/AKT ERK1/2 (in combination with lapatinib) | | GBM5,6,12 and 14 | [42] | |||
| | ||||||
| Human oesophageal carcinoma | OSU | Induces Apoptosis | BAX | | Eca-109, TE-1, and TE-11 cells | [43] |
| p-p53 | | |||||
| Cleaved caspase 3 | | |||||
| Cleaved caspase 9 | | |||||
| Cytochrome c release | | |||||
| Burkitt Lymphoma | DMC | Inhibits Cell cycle | Cyclin A/B | | Raji, Ramos and A6876 cells | [44] |
| P27Kip1 | | |||||
| Leukemia | DMC | Arrest of Cell cycle (G1/S) | c-myc | | U937, K562, Raji, Hel cells | [45] |
| Induces apoptosis | Survivin | | ||||
| Thyroid cancer | OSU | Inhibits cell cycle (S phase accumulation) | p-AKT | | NPA (papillary) WRO (follicular) ARO (anaplastic) cells | [46] |
| Induces apoptosis | Undetermined | - | ||||
| Migration/Invasion | PAK | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobolewski, C.; Legrand, N. Celecoxib Analogues for Cancer Treatment: An Update on OSU-03012 and 2,5-Dimethyl-Celecoxib. Biomolecules 2021, 11, 1049. https://doi.org/10.3390/biom11071049
Sobolewski C, Legrand N. Celecoxib Analogues for Cancer Treatment: An Update on OSU-03012 and 2,5-Dimethyl-Celecoxib. Biomolecules. 2021; 11(7):1049. https://doi.org/10.3390/biom11071049
Chicago/Turabian StyleSobolewski, Cyril, and Noémie Legrand. 2021. "Celecoxib Analogues for Cancer Treatment: An Update on OSU-03012 and 2,5-Dimethyl-Celecoxib" Biomolecules 11, no. 7: 1049. https://doi.org/10.3390/biom11071049
APA StyleSobolewski, C., & Legrand, N. (2021). Celecoxib Analogues for Cancer Treatment: An Update on OSU-03012 and 2,5-Dimethyl-Celecoxib. Biomolecules, 11(7), 1049. https://doi.org/10.3390/biom11071049