
Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants


 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Meta-Analysis
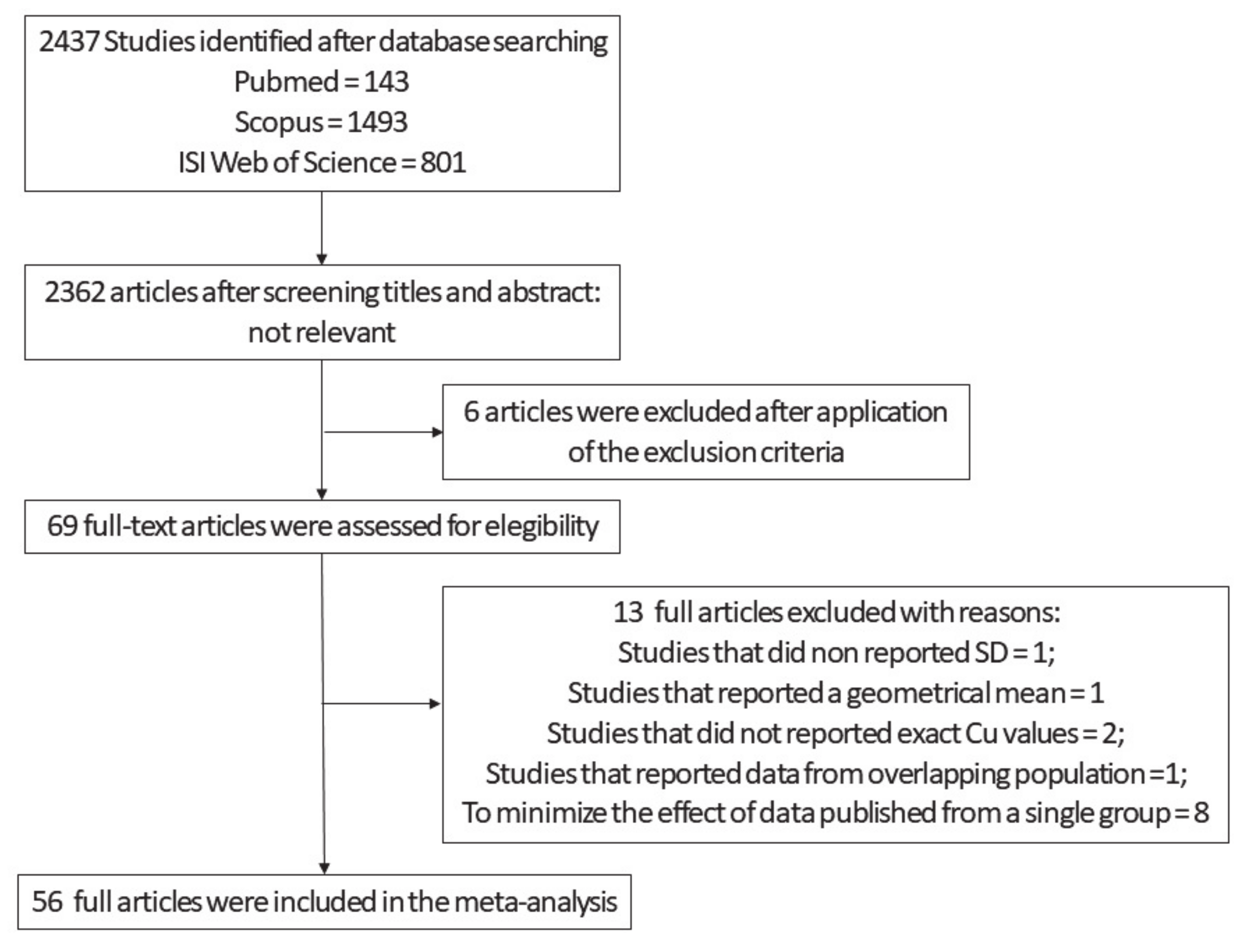
2.1.1. Search Strategy
2.1.2. Data Extraction and Manipulation for Meta-Analysis
2.1.3. Statistical Procedures Applied to Run Meta-Analysis
2.2. Replication Study Investigating Differences in Serum Cu Biomarkers between AD and HC
2.2.1. Subjects Analyzed in the Replication Study
2.2.2. Sample Collections
2.2.3. Biochemical Investigation Applied for the Replication Study
2.2.4. Statistical Analyses Applied to the Replication Study
2.2.5. Procedures Applied to Run the Genetic Studies
2.2.6. Statistical Analyses Applied for the Genetic Studies
3. Results
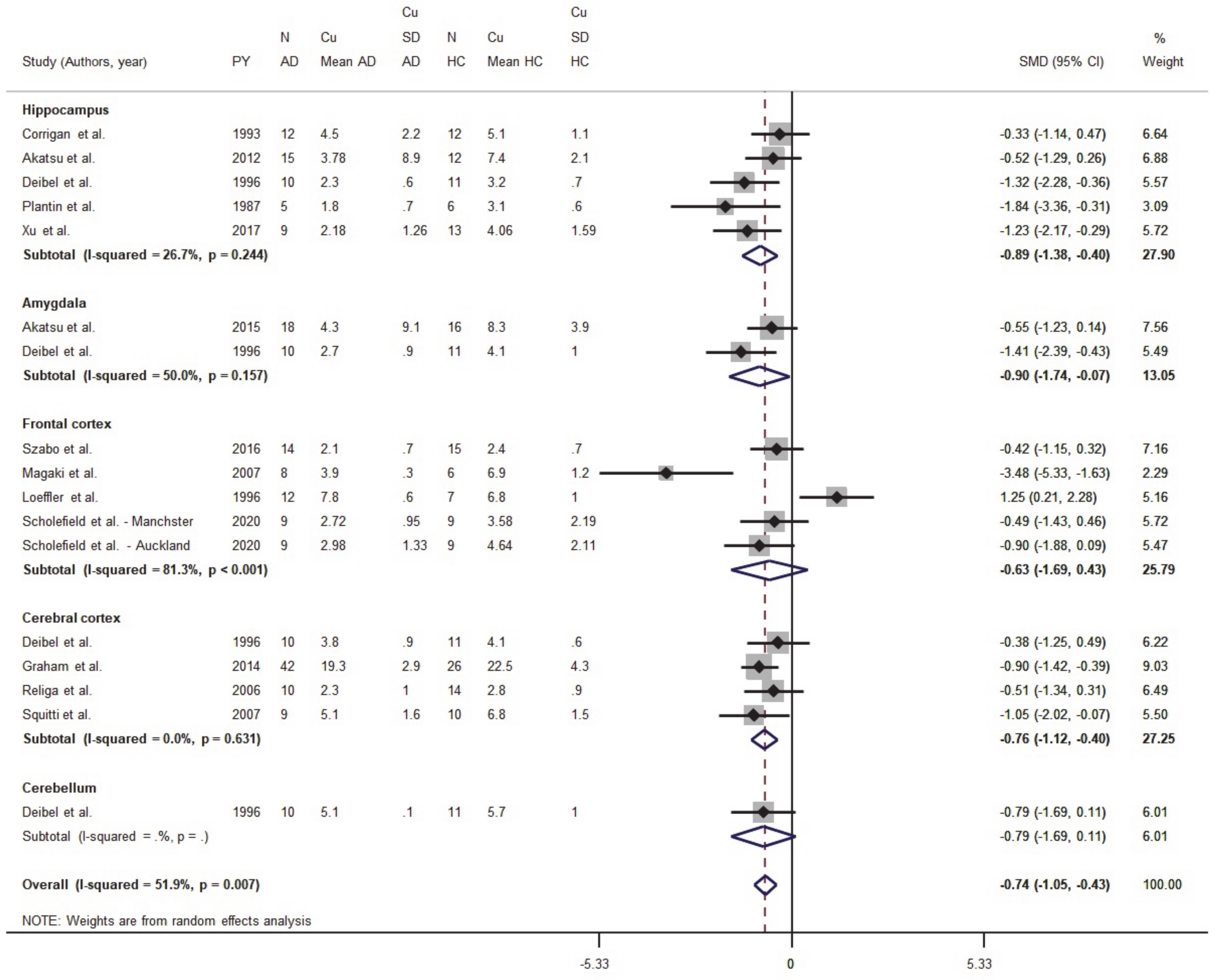
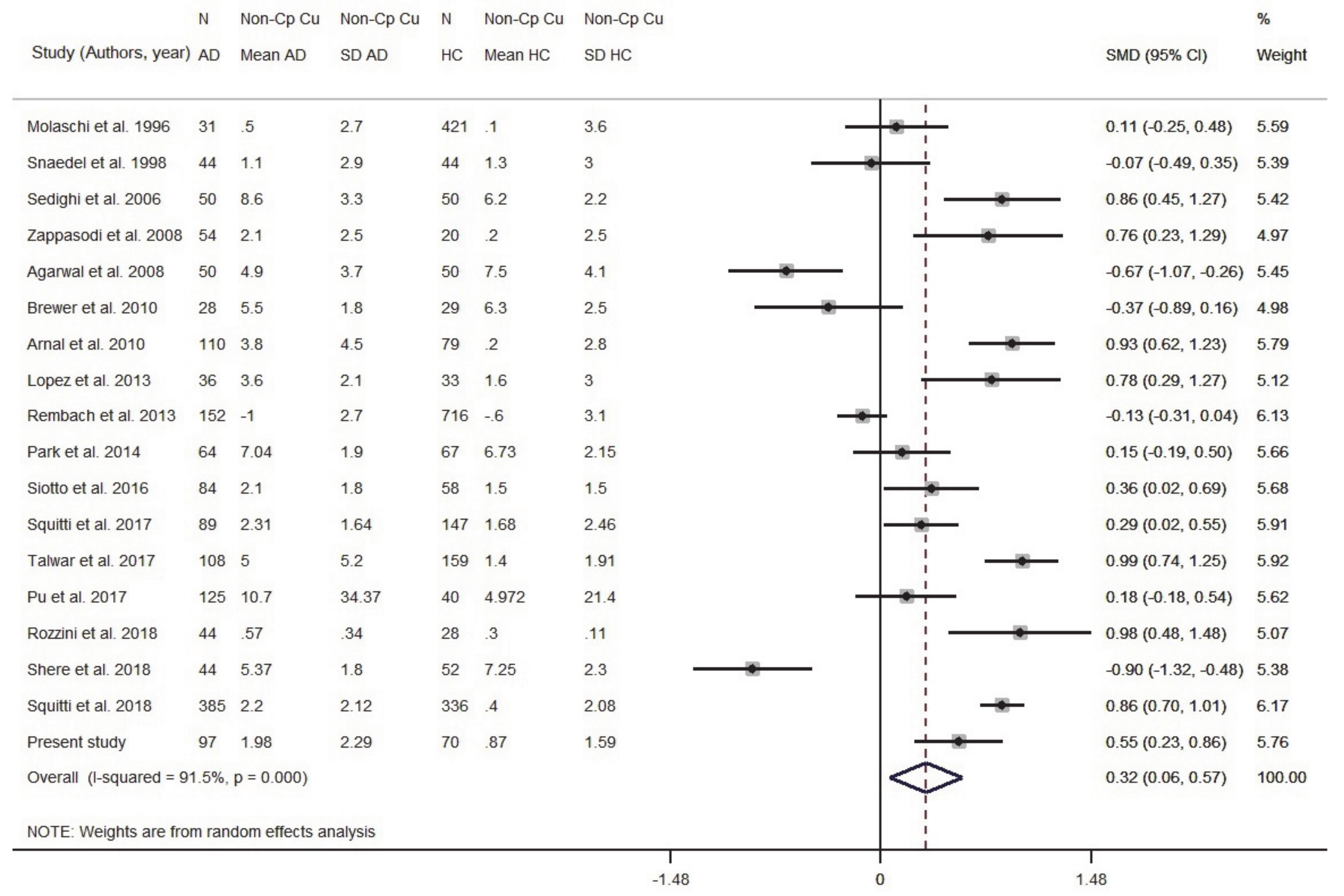
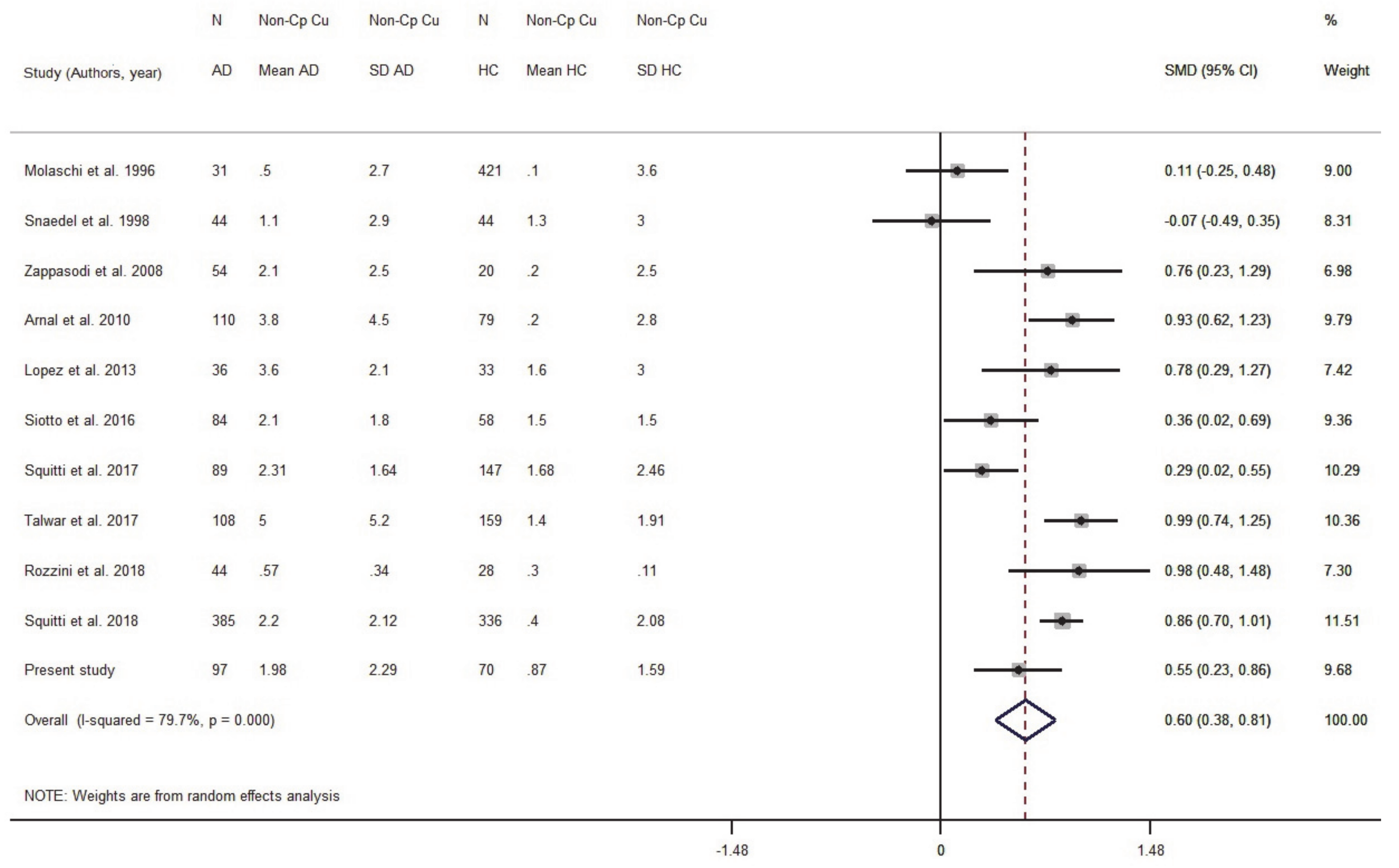
3.1. Meta-Analysis
3.1.1. Meta-Analysis of Cu Data from Brain Tissues
3.1.2. Meta-Analysis of Cu in Serum/Plasma
3.1.3. Meta-Analysis of Non-Cp Cu
3.1.4. Meta-Analysis of Ceruloplasmin in Serum/Plasma
3.2. Replication Study of Changes in Serum Cu Biomarkers in AD and HC Subjects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s-Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Kepp, K.P. Ten Challenges of the Amyloid Hypothesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- James, S.A.; Volitakis, I.; Adlard, P.A.; Duce, J.A.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 298–302. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Kepp, K.P.; Squitti, R. Copper imbalance in Alzheimer’s disease: Convergence of the chemistry and the clinic. Coord. Chem. Rev. 2019, 397, 168–187. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; la Penna, G. Metal ions and intrinsically disordered proteins and peptides: From Cu/Zn amyloid-beta to general principles. Acc. Chem. Res. 2014, 47, 2252–2259. [Google Scholar] [CrossRef]
- Hsu, H.W.; Rodriguez-Ortiz, C.J.; Lim, S.L.; Zumkehr, J.; Kilian, J.G.; Vidal, J.; Kitazawa, M. Copper-Induced Upregulation of MicroRNAs Directs the Suppression of Endothelial LRP1 in Alzheimer’s Disease Model. Toxicol. Sci. 2019, 170, 144–156. [Google Scholar] [CrossRef]
- Ma, Q.; Ying, M.; Sui, X.; Zhang, H.; Huang, H.; Yang, L.; Huang, X.; Zhuang, Z.; Liu, J.; Yang, X. Chronic copper exposure causes spatial memory impairment, selective loss of hippocampal synaptic proteins, and activation of PKR/eIF2alpha pathway in mice. J. Alzheimer’s Dis. 2015, 43, 1413–1427. [Google Scholar] [CrossRef]
- Wu, M.; Han, F.; Gong, W.; Feng, L.; Han, J. The effect of copper from water and food: Changes of serum nonceruloplasmin copper and brain’s amyloid-beta in mice. Food Funct. 2016, 7, 3740–3747. [Google Scholar] [CrossRef]
- Yu, J.; Luo, X.; Xu, H.; Ma, Q.; Yuan, J.; Li, X.; Chang, R.C.; Qu, Z.; Huang, X.; Zhuang, Z.; et al. Identification of the key molecules involved in chronic copper exposure-aggravated memory impairment in transgenic mice of Alzheimer’s disease using proteomic analysis. J. Alzheimer’s Dis. 2015, 44, 455–469. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, zinc and copper in the Alzheimer’s disease brain: A quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-D.; Zhang, W.; Wang, Z.-Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci. 2017, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Simonelli, I.; Ventriglia, M.; Siotto, M.; Pasqualetti, P.; Rembach, A.; Doecke, J.; Bush, A.I. Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. J Alzheimers Dis. 2014, 38, 809–822. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Zabel, M.; Crofton, A.; Kirsch, W.M.; Ghribi, O.; Squitti, R.; Perry, G. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Neurobiol. Dis. 2013, 59, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Polimanti, R.; Bucossi, S.; Ventriglia, M.; Mariani, S.; Manfellotto, D.; Vernieri, F.; Cassetta, E.; Ursini, F.; Rossini, P.M. Linkage disequilibrium and haplotype analysis of the ATP7B gene in Alzheimer’s disease. Rejuvenation Res. 2013, 16, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, C.J.; Jayakanthan, S.; Siotto, M.; Yang, N.; Osipova, M.; Squitti, R.; Lutsenko, S. Single nucleotide polymorphisms in the human ATP7B gene modify the properties of the ATP7B protein. Metallomics 2019, 11, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Hozo, S.P.; Djulbegovic, B.; Hozo, I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med. Res. Methodol. 2005, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Wang, W.; Liu, J.; Tong, T. Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Med. Res. Methodol. 2014, 14, 135. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.; Thomas, J.; Chandler, J.; Cumpston, M.; Li, T.; Page, M.; Welch, V.A. Cochrane Handbook for Systematic Reviews of Interventions Version 6.0; (Updated July 2019); Cochrane: London, UK, 2019; Available online: www.training.cochrane.org/handbook (accessed on 28 June 2021).
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Cummings, Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Squitti, R.; Pasqualetti, P.; Forno, G.D.; Moffa, F.; Cassetta, E.; Lupoi, D.; Vernieri, F.; Rossi, L.; Baldassini, M.; Rossini, P.M. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology 2005, 64, 1040–1046. [Google Scholar] [CrossRef]
- Wolf, P.L. Ceruloplasmin: Methods and clinical use. Crit. Rev. Clin. Lab. Sci. 1982, 17, 229–245. [Google Scholar] [CrossRef]
- Schosinsky, K.H.; Lehmann, H.P.; Beeler, M.F. Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin. Chem. 1974, 20, 1556–1563. [Google Scholar] [CrossRef]
- Siotto, M.; Pasqualetti, P.; Marano, M.; Squitti, R. Automation of o-dianisidine assay for ceruloplasmin activity analyses: Usefulness of investigation in Wilson’s disease and in hepatic encephalopathy. J. Neural Transm. 2014, 121, 1281–1286. [Google Scholar] [CrossRef]
- Abe, A.; Yamashita, S.; Noma, A. Sensitive, direct colorimetric assay for copper in serum. Clin. Chem. 1989, 35, 552–554. [Google Scholar] [CrossRef]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M.; et al. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef]
- Walshe, J.M.B. Clinical Investigations Standing Committee of the Association of Clinical, Wilson’s disease: The importance of measuring serum caeruloplasmin non-immunologically. Ann. Clin. Biochem. 2003, 40, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Twomey, P.J.; Viljoen, A.; House, I.M.; Reynolds, T.M.; Wierzbicki, A.S. Copper: Caeruloplasmin ratio. J. Clin. Pathol. 2007, 60, 441–442. [Google Scholar] [CrossRef] [Green Version]
- Ward, N.I.; Mason, J.A. Neutron activation analysis techniques for identifying elemental status in Alzheimer’s disease. J. Radioanal. Nucl. Chem. 1987, 113, 515–526. [Google Scholar] [CrossRef]
- McIntosh, K.G.; Cusack, M.J.; Vershinin, A.; Chen, Z.W.; Zimmerman, E.A.; Molho, E.S.; Celmins, D.; Parsons, P.J. Evaluation of a prototype point-of-care instrument based on monochromatic X-ray fluorescence spectrometry: Potential for monitoring trace element status of subjects with neurodegenerative disease. J. Toxicol. Environ. Health A 2012, 75, 1253–1268. [Google Scholar] [CrossRef]
- Mueller, C.; Schrag, M.; Crofton, A.; Stolte, J.; Muckenthaler, M.U.; Magaki, S.; Kirsch, W. Altered Serum Iron and Copper Homeostasis Predicts Cognitive Decline in Mild Cognitive Impairment. J. Alzheimer’s Dis. 2012, 29, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Alimonti, A.; Ristori, G.; Giubilei, F.; Stazi, M.A.; Pino, A.; Visconti, A.; Brescianini, S.; Monti, M.S.; Forte, G.; Stanzione, P.; et al. Serum chemical elements and oxidative status in Alzheimer’s disease, Parkinson disease and multiple sclerosis. Neurotoxicology 2007, 28, 450–456. [Google Scholar] [CrossRef]
- Bocca, B.; Forte, G.; Petrucci, F.; Pino, A.; Marchione, F.; Bomboi, G.; Senofonte, O.; Giubilei, F.; Alimonti, A. Monitoring of chemical elements and oxidative damage in patients affected by Alzheimer’s disease. Ann. Ist. Super Sanita 2005, 41, 197–203. [Google Scholar]
- Squitti, R.; Siotto, M.; Cassetta, E.; Idrissi, I.G.; Colabufo, N.A. Measurements of serum non-ceruloplasmin copper by a direct fluorescent method specific to Cu(II). Clin. Chem. Lab. Med. 2017, 55, 1360–1367. [Google Scholar] [CrossRef]
- Siotto, M.; Simonelli, I.; Pasqualetti, P.; Mariani, S.; Caprara, D.; Bucossi, S.; Ventriglia, M.; Molinario, R.; Antenucci, M.; Rongioletti, M.; et al. Association Between Serum Ceruloplasmin Specific Activity and Risk of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 50, 1181–1189. [Google Scholar] [CrossRef]
- Zappasodi, F.; Salustri, C.; Babiloni, C.; Cassetta, E.; del Percio, C.; Ercolani, M.; Rossini, P.M.; Squitti, R. An observational study on the influence of the APOE-epsilon4 allele on the correlation between ‘free’ copper toxicosis and EEG activity in Alzheimer disease. Brain Res. 2008, 1215, 183–189. [Google Scholar] [CrossRef]
- Squitti, R.; Lupoi, D.; Pasqualetti, P.; Forno, G.D.; Vernieri, F.; Chiovenda, P.; Rossi, L.; Cortesi, M.; Cassetta, E.; Rossini, P.M. Elevation of serum copper levels in Alzheimer’s disease. Neurology 2002, 59, 1153–1161. [Google Scholar] [CrossRef]
- Plantin, L.O.; Lying-Tunell, U.; Kristensson, K. Trace elements in the human central nervous system studied with neutron activation analysis. Biol. Trace Elem. Res. 1987, 13, 69–75. [Google Scholar] [CrossRef]
- Corrigan, F.M.; Reynolds, G.P.; Ward, N.I. Hippocampal tin, aluminum and zinc in Alzheimer’s disease. Biometals 1993, 6, 149–154. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Le Witt, P.A.; Juneau, P.L.; Sima, A.A.; Nguyen, H.U.; De Maggio, A.J.; Brickman, C.M.; Brewer, G.J.; Dick, R.D.; Troyer, M.D.; et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 1996, 738, 265–274. [Google Scholar] [CrossRef]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Squitti, R.; Quattrocchi, C.C.; Forno, G.D.; Antuono, P.; Wekstein, D.R.; Capo, C.R.; Salustri, C.; Rossini, P.M. Ceruloplasmin (2-D PAGE) Pattern and Copper Content in Serum and Brain of Alzheimer Disease Patients. Biomark Insights 2007, 1, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Religa, D.; Strozyk, D.; Cherny, R.A.; Volitakis, I.; Haroutunian, V.; Winblad, B.; Naslund, J.; Bush, A.I. Elevated cortical zinc in Alzheimer disease. Neurology 2006, 67, 69–75. [Google Scholar] [CrossRef]
- Magaki, S.; Raghavan, R.; Mueller, C.; Oberg, K.C.; Vinters, H.V.; Kirsch, W.M. Iron, copper, and iron regulatory protein 2 in Alzheimer’s disease and related dementias. Neurosci. Lett. 2007, 418, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akatsu, H.; Hori, A.; Yamamoto, T.; Yoshida, M.; Mimuro, M.; Hashizume, Y.; Tooyama, I.; Yezdimer, E.M. Transition metal abnormalities in progressive dementias. Biometals 2012, 25, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Nasaruddin, M.B.; Carey, M.; Holscher, C.; McGuinness, B.; Kehoe, P.G.; Love, S.; Passmore, P.; Elliott, C.T.; Meharg, A.A.; et al. Age-associated changes of brain copper, iron, and zinc in Alzheimer’s disease and dementia with Lewy bodies. J. Alzheimer’s Dis. 2014, 42, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.T.; Harry, G.J.; Hayden, K.M.; Szabo, D.T.; Birnbaum, L. Comparison of Metal Levels between Postmortem Brain and Ventricular Fluid in Alzheimer’s Disease and Nondemented Elderly Controls. Toxicol. Sci. 2016, 150, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 2017, 9, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholefield, M.; Church, S.J.; Xu, J.; Kassab, S.; Gardiner, N.J.; Roncaroli, F.; Hooper, N.M.; Unwin, R.D.; Cooper, G.J.S. Evidence that levels of nine essential metals in post-mortem human-Alzheimer’s-brain and ex vivo rat-brain tissues are unaffected by differences in post-mortem delay, age, disease staging, and brain bank location. Metallomics 2020, 12, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Andrasi, E.; Farkas, E.; Scheibler, H.; Reffy, A.; Bezur, L. Al, Zn, Cu, Mn and Fe levels in brain in Alzheimer’s disease. Arch. Gerontol. Geriatr. 1995, 21, 89–97. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Rao, K.; Rao, R.; Shanmugavelu, P.; Menon, R. Trace elements in Alzheimer’s disease: A new hypothesis. Alzheimer’s Rep. 1999, 2, 241–246. [Google Scholar]
- Tandon, L.; Ni, B.F.; Ding, X.X.; Ehmann, W.D.; Kasarskis, E.J.; Markesbery, W.R. RNAA for arsenic, cadmium, copper and molybdenum in CNS tissue from subjects with age-related neurodegenerative diseases. J. Radioanal. Nucl. Res. 1994, 179, 331–339. [Google Scholar] [CrossRef]
- House, E.; Esiri, M.; Forster, G.; Ince, P.G.; Exley, C. Aluminium, iron and copper in human brain tissues donated to the Medical Research Council’s Cognitive Function and Ageing Study. Metallomics 2012, 4, 56–65. [Google Scholar] [CrossRef]
- Yoshimasu, F.; Yasui, M.; Yase, Y.; Iwata, S.; Gajdusek, D.C.; Gibbs, C.J., Jr.; Chen, J.K.M. Studies on amyotrophic lateral sclerosis by neutron activation analysis-2. Comparative study of analytical results on Guam PD, Japanese ALS and Alzheimer disease cases. Folia Psychiatr. Neurol. Jpn. 1980, 34, 75–82. [Google Scholar] [CrossRef]
- Gonzalez, C.; Martin, T.; Cacho, J.; Brenas, M.T.; Arroyo, T.; Garcia-Berrocal, B.; Navajo, J.A.; Gonzalez-Buitrago, J.M. Serum zinc, copper, insulin and lipids in Alzheimer’s disease epsilon 4 apolipoprotein E allele carriers. Eur. J. Clin. Investig. 1999, 29, 637–642. [Google Scholar] [CrossRef]
- Smorgon, C.; Mari, E.; Atti, A.R.; Nora, E.D.; Zamboni, P.F.; Calzoni, F.; Passaro, A.; Fellin, R. Trace elements and cognitive impairment: An elderly cohort study. Arch. Gerontol. Geriatr. 2004, 38, 393–402. [Google Scholar] [CrossRef]
- Sevym, S.; Ozgur, U.; Tamer, L.; Okan, D.; Aynur, O. Can serum levels of copper and zinc distinguish Alzheimer’s patients from normal subjects? J. Neurol. Sci. 2007, 24, 197–205. [Google Scholar]
- Agarwal, R.; Kushwaha, S.S.; Tripathi, C.B.; Singh, N.; Chhillar, N. Serum copper in Alzheimer’s disease and vascular dementia. Indian J. Clin. Biochem. 2008, 23, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Azhdarzadeh, M.; Noroozian, M.; Aghaverdi, H.; Akbari, S.M.; Baum, L.; Mahmoudi, M. Serum multivalent cationic pattern: Speculation on the efficient approach for detection of Alzheimer’s disease. Sci. Rep. 2013, 3, 2782. [Google Scholar] [CrossRef] [Green Version]
- Lopez, N.; Tormo, C.; de Blas, I.; Llinares, I.; Alom, J. Oxidative stress in Alzheimer’s disease and mild cognitive impairment with high sensitivity and specificity. J. Alzheimer’s Dis. 2013, 33, 823–829. [Google Scholar] [CrossRef]
- Singh, N.K.; Banerjee, B.D.; Bala, K.; Basu, M.; Chhillar, N. Polymorphism in Cytochrome P450 2D6, Glutathione S-Transferases Pi 1 Genes, and Organochlorine Pesticides in Alzheimer Disease: A Case-Control Study in North Indian Population. J. Geriatr. Psychiatry Neurol. 2014, 27, 119–127. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, D.W.; Park, K.S. Elevated serum copper and ceruloplasmin levels in Alzheimer’s disease. Asia Pac. Psychiatry 2014, 6, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Miedico, O.; Cristofano, A.; Vitale, M.; Angiolillo, A.; Chiaravalle, A.E.; Corso, G.; di Costanzo, A. Distinctive Pattern of Serum Elements During the Progression of Alzheimer’s Disease. Sci. Rep. 2016, 6, 22769. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Tan, L.; Wang, H.F.; Ma, J.; Liu, J.; Tan, M.S.; Sun, J.H.; Zhu, X.C.; Jiang, T.; Yu, J.T. Serum Iron, Zinc, and Copper Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analyses. J. Alzheimer’s Dis. 2015, 47, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Grover, S.; Sinha, J.; Chandna, P.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Multifactorial Analysis of a Biomarker Pool for Alzheimer Disease Risk in a North Indian Population. Dement. Geriatr. Cogn. Disord. 2017, 44, 25–34. [Google Scholar] [CrossRef]
- Rozzini, L.; Lanfranchi, F.; Pilotto, A.; Catalani, S.; Gilberti, M.E.; Paganelli, M.; Apostoli, P.; Padovani, A. Serum Non-Ceruloplasmin Non-Albumin Copper Elevation in Mild Cognitive Impairment and Dementia due to Alzheimer’s Disease: A Case Control Study. J. Alzheimer’s Dis. 2018, 61, 907–912. [Google Scholar] [CrossRef]
- Rembach, A.; Doecke, J.D.; Roberts, B.R.; Watt, A.D.; Faux, N.G.; Volitakis, I.; Pertile, K.K.; Rumble, R.L.; Trounson, B.O.; Fowler, C.J.; et al. Longitudinal analysis of serum copper and ceruloplasmin in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 34, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Shere, S.; Subramanian, S.; Bharath, S.; Purushottam, M. Lower levels of serum copper in patients with Alzheimer’s dementia: A controlled study from India. Asian J. Psychiatr. 2018, 32, 73–74. [Google Scholar] [CrossRef]
- Basun, H.; Forssell, L.G.; Wetterberg, L.; Winblad, B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Park Dis. Dement. Sect. 1991, 3, 231–258. [Google Scholar]
- Mattiello, G.; Gerotto, M.; Favarato, M.; Lazzri, F.; Gasparoni, G.; Gomirato, L.; Mazzolini, G.; Scarpa, G.; Zanaboni, V.; Pilone, M.G.; et al. Plasma Microelement Analysis from Alzheimer’s and Multi-infartual Dementia Patients. Alzheimer’s Dis. Adv. Clin. Basic Res. 1993, 267–272. [Google Scholar]
- Ashraf, A.; Stosnach, H.; Parkes, H.G.; Hye, A.; Powell, J.; So, P.-W.; AddNeuroMed Consortium. Pattern of Altered Plasma Elemental Phosphorus, Calcium, Zinc, and Iron in Alzheimer’s Disease. Sci. Rep. 2019, 9, 3147. [Google Scholar] [CrossRef]
- Arnal, N.; Cristalli, D.O.; de Alaniz, M.J.; Marra, C.A. Clinical utility of copper, ceruloplasmin, and metallothionein plasma determinations in human neurodegenerative patients and their first-degree relatives. Brain Res. 2010, 1319, 118–130. [Google Scholar] [CrossRef]
- Alsadany, M.A.; Shehata, H.H.; Mohamad, M.I.; Mahfouz, R.G. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Demen. 2013, 28, 54–61. [Google Scholar] [CrossRef]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Kellett, K.A.B.; Vardy, E.; Unwin, R.D.; Hooper, N.M.; Cooper, G.J.S. Plasma metals as potential biomarkers in dementia: A case-control study in patients with sporadic Alzheimer’s disease. Biometals 2018, 31, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Giacconi, R.; Giuli, C.; Casoli, T.; Balietti, M.; Costarelli, L.; Provinciali, M.; Basso, A.; Piacenza, F.; Postacchini, D.; Galeazzi, R.; et al. Acetylcholinesterase inhibitors in Alzheimer’s disease influence Zinc and Copper homeostasis. J. Trace Elem. Med. Biol. 2019, 55, 58–63. [Google Scholar] [CrossRef]
- Vural, H.; Demirin, H.; Kara, Y.; Eren, I.; Delibas, N. Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. J. Trace Elem. Med. Biol. 2010, 24, 169–173. [Google Scholar] [CrossRef]
- Gerhardsson, L.; Lundh, T.; Minthon, L.; Londos, E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 25, 508–515. [Google Scholar] [CrossRef]
- Roberts, E.A.; Schilsky, M.L. American Association for Study of Liver Diseases. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef]
- European Association for Study of the Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Reed, E.; Lutsenko, S.; Bandmann, O. Animal models of Wilson disease. J. Neurochem. 2018, 146, 356–373. [Google Scholar] [CrossRef]
- Fujiwara, N.; Iso, H.; Kitanaka, N.; Kitanaka, J.; Eguchi, H.; Ookawara, T.; Ozawa, K.; Shimoda, S.; Yoshihara, D.; Takemura, M.; et al. Effects of copper metabolism on neurological functions in Wistar and Wilson’s disease model rats. Biochem. Biophys. Res. Commun. 2006, 349, 1079–1086. [Google Scholar] [CrossRef]
- Squitti, R.; Ventriglia, M.; Gennarelli, M.; Colabufo, N.A.; el Idrissi, I.G.; Bucossi, S.; Mariani, S.; Rongioletti, M.; Zanetti, O.; Congiu, C.; et al. Non-Ceruloplasmin Copper Distincts Subtypes in Alzheimer’s Disease: A Genetic Study of ATP7B Frequency. Mol. Neurobiol. 2017, 54, 671–681. [Google Scholar] [CrossRef]
- Squitti, R.; Ghidoni, R.; Siotto, M.; Ventriglia, M.; Benussi, L.; Paterlini, A.; Magri, M.; Binetti, G.; Cassetta, E.; Caprara, D.; et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann. Neurol. 2014, 75, 574–580. [Google Scholar] [CrossRef]
- Squitti, R.; Pasqualetti, P.; Polimanti, R.; Salustri, C.; Moffa, F.; Cassetta, E.; Lupoi, D.; Ventriglia, M.; Cortesi, M.; Siotto, M.; et al. Metal-score as a potential non-invasive diagnostic test for Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 191–198. [Google Scholar] [CrossRef]
- Squitti, R.; Bressi, F.; Pasqualetti, P.; Bonomini, C.; Ghidoni, R.; Binetti, G.; Cassetta, E.; Moffa, F.; Ventriglia, M.; Vernieri, F.; et al. Longitudinal prognostic value of serum “free” copper in patients with Alzheimer disease. Neurology 2009, 72, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-beta homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef] [Green Version]
- Coelho, F.C.; Squitti, R.; Ventriglia, M.; Cerchiaro, G.; Daher, J.P.; Rocha, J.G.; Rongioletti, M.C.A.; Moonen, A.C. Agricultural Use of Copper and Its Link to Alzheimer’s Disease. Biomolecules 2020, 10, 897. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.; Harris, C.; Ralle, M.; Duffy, M.; Murchison, C.; Quinn, J.F. Modulation of tau phosphorylation by environmental copper. Transl. Neurodegener. 2014, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council. Copper in Drinking Water; The National Academies Press: Washington, DC, USA, 2020; 162p. [Google Scholar] [CrossRef]
- Barnard, N.D.; Bush, A.I.; Ceccarelli, A.; Cooper, J.; de Jager, C.A.; Erickson, K.I.; Fraser, G.; Kesler, S.; Levin, S.M.; Lucey, B.; et al. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol. Aging 2014, 35 (Suppl. 2), S74–S78. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Siotto, M.; Polimanti, R. Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol. Aging 2014, 35 (Suppl. 2), S40–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Diouf, I.; Bush, A.I.; Ayton, S.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid ceruloplasmin levels predict cognitive decline and brain atrophy in people with underlying beta-amyloid pathology. Neurobiol. Dis. 2020, 139, 104810. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study (Authors, Year) | Country | Brain Region | Meta-Analysis Classification | Alzheimer’s Dementia | Healthy Controls | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Sex (F) | Mean Age (SD) | Cu μg/g (SD) | N | Sex (F) | Mean Age (SD) | Cu μg/g (SD) | ||||
| Plantin et al., 1987 [47] | Sweden | temporal lobe | hippocampus | 5 | 1.8 (0.7) | 6 | 3.1 (0.6) | ||||
| Corrigan et al., 1993 [48] | UK | hippocampal tissue | hippocampus | 12 | 10 | 79.5 (9.2) | 4.5 (2.2) | 12 | 4 | 78.5 (9.0) | 5.1 (1.1) |
| Loeffler et al., 1996 [49] | USA | frontal cortex | frontal cortex | 12 | 79.4 (1.7) | 7.8 (0.6) | 7 | 75.7 (2.8) | 6.8 (1.0) | ||
| Amygdala | Amygdala | 10 | 3 | 80.4 | 2.7 (0.9) | 11 | 8 | 81.7 | 4.1 (1.0) | ||
| Hippocampus | hippocampus | 2.3 (0.6) | 3.2 (0.7) | ||||||||
| Deibel et al., 1996 [50] | USA | superior and middle temporal | cerebral cortex | 3.2 (0.6) | 4.0 (0.6) | ||||||
| inferior parietal | cerebral cortex | 3.8 (0.9) | 4.1 (0.6) | ||||||||
| Cerebellum | Cerebellum | 5.1 (0.1) | 5.7 (1.0) | ||||||||
| Squitti et al., 2007 [51] | Italian | cortical tissue | cerebral cortex | 9 | 84.6 | 5.1 (1.6) | 10 | 80.2 (6.8) | 6.8 (1.5) | ||
| Religa et al., 2006 [52] | Australia | neocortical tissue | cerebral cortex | 10 | 7 | 81.6 (11) | 2.3 (1.0) | 14 | 12 | 82.8 (11.2) | 2.8 (0.9) |
| Magaki et al., 2007 [53] | USA | frontal cortex | frontal cortex | 8 | 6 | 78 (12) | 3.9 (0.3) | 6 | 2 | 72 (11.0) | 6.9 (1.2) |
| Akatsu et al., 2012 [54] | Japan | Hippocampus | hippocampus | 15 | 9 | 87.5 (7.6) | 3.78 (8.9) | 12 | 6 | 84 (6.7) | 7.4 (2.1) |
| Amygdala | Amygdala | 18 | 11 | 4.3 (9.1) | 16 | 10 | 85.9 (7.3) | 8.3 (3.9) | |||
| Graham et al., 2014 [55] | UK | Brodman area 7 | cerebral cortex | 42 | 21 | 83 (7.2) | 19.3 (2.9) | 26 | 13 | 81.7 (6.2) | 22.5 (4.3) |
| Szabo et al., 2016 [56] | USA | frontal cortex | frontal cortex | 14 | 78 | 2.1 (0.7) | 15 | 88 | 2.4 (0.7) | ||
| Xu et al., 2017 [57] | New Zealand | Hippocampus | hippocampus | 9 | 4 | 72 (60–80) | 2.18 (1.26) | 13 | 5 | 73 (61–78) | 4.06 (1.59) |
| Scholefield et al., 2020 [58] | UK | Cingulate gyrus | frontal cortex | 9 | 6 | 83 (61–89) | 2.72 (0.95) | 9 | 3 | 89 (82–95) | 3.58 (2.19) |
| Scholefield et al., 2020 [58] | New Zealand | Cingulate gyrus | frontal cortex | 9 | 4 | 72 (60–80) | 2.98 (1.33) | 9 | 2 | 73 (61–78) | 4.64 (2.11) |
| Alzheimer’s Dementia | Healthy Controls | Statistics | p-Value | |||
|---|---|---|---|---|---|---|
| N = 97 | N = 70 | |||||
| Demographic variables | ||||||
| Sex, M | % (n/subjects) | 37.1% (36/97) | 22.9% (16/70) | 0.05 | ||
| Age | mean (SD) | 71.1 (7.18) | 65.9 (7.53) | 0.01 | ||
| MMSE | median (Q1-Q3) | 19 (15–23) | 29 (27.3–30) | <0.001 * | ||
| Biological variables | ||||||
| Cu (µmol/L) | mean (SD) | 15.8 (3.76) | 14.6 (2.95) | F(1, 163) = 6.69 # | 0.011 | |
| Ceruloplasmin (mg/dL) | mean (SD) | 29.3 (4.89) | 29.2 (4.08) | F(1, 163) = 1.13 # | 0.289 | |
| Non-ceruloplasmin Cu *,&,† (µmol/L) | median(Q1-Q3) | 1.4 (0.6–2.8) | 0.75 (−0.16–1.71) | F(1, 163) = 9.88 # | 0.002 | |
| Ceruloplasmin activity (IU) | mean (SD) | 117.9 (26.13) | 110.4 (18.06) | F(1, 163) = 10.21 # | 0.008 | |
| eCp:iCp ratio | mean (SD) | 4.0 (0.51) | 3.8 (0.44) | F(1, 163) = 7.02 # | 0.009 | |
| Cu:Cp ratio | mean (SD) | 7.1 (0.74) | 6.6 (0.93) | F(1, 163) = 10.21 # | 0.002 | |
| Healthy Controls | AD | Chi-Square | Odd Ratio | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs732774 | n | Freq | n | Freq | Value | df | p | pcorrect | Value | 95% CI | |
| Alleles | |||||||||||
| A | 78 | 0.57 | 113 | 0.62 | |||||||
| G | 60 | 0.43 | 69 | 0.38 | 1.011 | 1 | 0.315 | 0.409 | |||
| Total | 138 | 1.00 | 182 | 1.00 | |||||||
| Genotypes | |||||||||||
| AA | 24 | 0.35 | 29 | 0.32 | |||||||
| AG | 30 | 0.43 | 55 | 0.60 | 7.857 | 2 | 0.020 | 0.032 | |||
| GG | 15 | 0.22 | 7 | 0.08 | |||||||
| Total | 69 | 1.00 | 91 | 1.00 | |||||||
| Carriers | |||||||||||
| Allele A | 54 | 0.78 | 84 | 0.92 | 6.529 | 1 | 0.011 | 0.011 | 3.33 | 1.28 | 8.70 |
| Allele G | 45 | 0.65 | 62 | 0.68 | 0.151 | 1 | 0.698 | 0.799 | 1.14 | 0.59 | 2.21 |
| Healthy Controls | AD | Chi-Square | Odd Ratio | ||||||||
| rs1061472 | N | Freq | N | Freq | Value | df | p | pcorrect | Value | 95% CI | |
| Alleles | |||||||||||
| A | 64 | 0.46 | 72 | 0.40 | |||||||
| G | 74 | 0.54 | 110 | 0.60 | 1.492 | 1 | 0.222 | 0.315 | |||
| Total | 138 | 1.00 | 182 | 1.00 | |||||||
| Genotypes | |||||||||||
| AA | 17 | 0.25 | 10 | 0.11 | |||||||
| AG | 30 | 0.43 | 52 | 0.57 | 5.762 | 2 | 0.056 | 0.097 | |||
| GG | 22 | 0.32 | 29 | 0.32 | |||||||
| Total | 69 | 1.00 | 92 | 1.00 | |||||||
| Carriers | |||||||||||
| Allele A | 47 | 0.68 | 62 | 0.68 | 4.6 × 10−6 | 1 | 0.998 | 1.000 | 1.00 | 0.51 | 1.96 |
| Allele G | 52 | 0.75 | 81 | 0.89 | 5.212 | 1 | 0.022 | 0.031 | 2.65 | 1.13 | 6.21 |
| Univariable Analysis | Multivariable Analysis * | |||||
|---|---|---|---|---|---|---|
| OR | 95% CI | p-Value | OR | 95% CI | p-Value | |
| Sex (M vs. F) | 1.99 | 0.99–3.98 | 0.051 | 2.51 | 1.13–5.56 | 0.023 |
| Age | 1.06 | 1.01–1.1 | 0.008 | 1.03 | 0.98–1.08 | 0.193 |
| Cu | 1.11 | 1.01–1.22 | 0.037 | |||
| iCp | 1.01 | 0.94–1.08 | 0.845 | |||
| eCp | 1.02 | 1.0–1.032 | 0.045 | |||
| Non-Cp Cu | 1.4 | 1.14–1.70 | 0.001 | 1.32 | 1.06–1.64 | 0.012 |
| eCp:iCp ratio | 2.86 | 1.36–6.0 | 0.006 | 2.17 | 0.96–4.91 | 0.062 |
| Cu:Cp ratio | 1.17 | 0.46–3.01 | 0.74 | |||
| Haplotypes * | ||||||
| AG vs. GA/GA | 2.7 | 1.13–6.23 | 0.026 | 2.49 | 0.99–6.28 | 0.053 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules 2021, 11, 960. https://doi.org/10.3390/biom11070960
Squitti R, Ventriglia M, Simonelli I, Bonvicini C, Costa A, Perini G, Binetti G, Benussi L, Ghidoni R, Koch G, et al. Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules. 2021; 11(7):960. https://doi.org/10.3390/biom11070960
Chicago/Turabian StyleSquitti, Rosanna, Mariacarla Ventriglia, Ilaria Simonelli, Cristian Bonvicini, Alfredo Costa, Giulia Perini, Giuliano Binetti, Luisa Benussi, Roberta Ghidoni, Giacomo Koch, and et al. 2021. "Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants" Biomolecules 11, no. 7: 960. https://doi.org/10.3390/biom11070960
APA StyleSquitti, R., Ventriglia, M., Simonelli, I., Bonvicini, C., Costa, A., Perini, G., Binetti, G., Benussi, L., Ghidoni, R., Koch, G., Borroni, B., Albanese, A., Sensi, S. L., & Rongioletti, M. (2021). Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules, 11(7), 960. https://doi.org/10.3390/biom11070960