
Determination of Butyrate Synthesis Capacity in Gut Microbiota: Quantification of but Gene Abundance by qPCR in Fecal Samples

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Description of the Study Population
2.2. Fecal Samples Handling and Storage
2.3. DNA Isolation from Fecal Samples
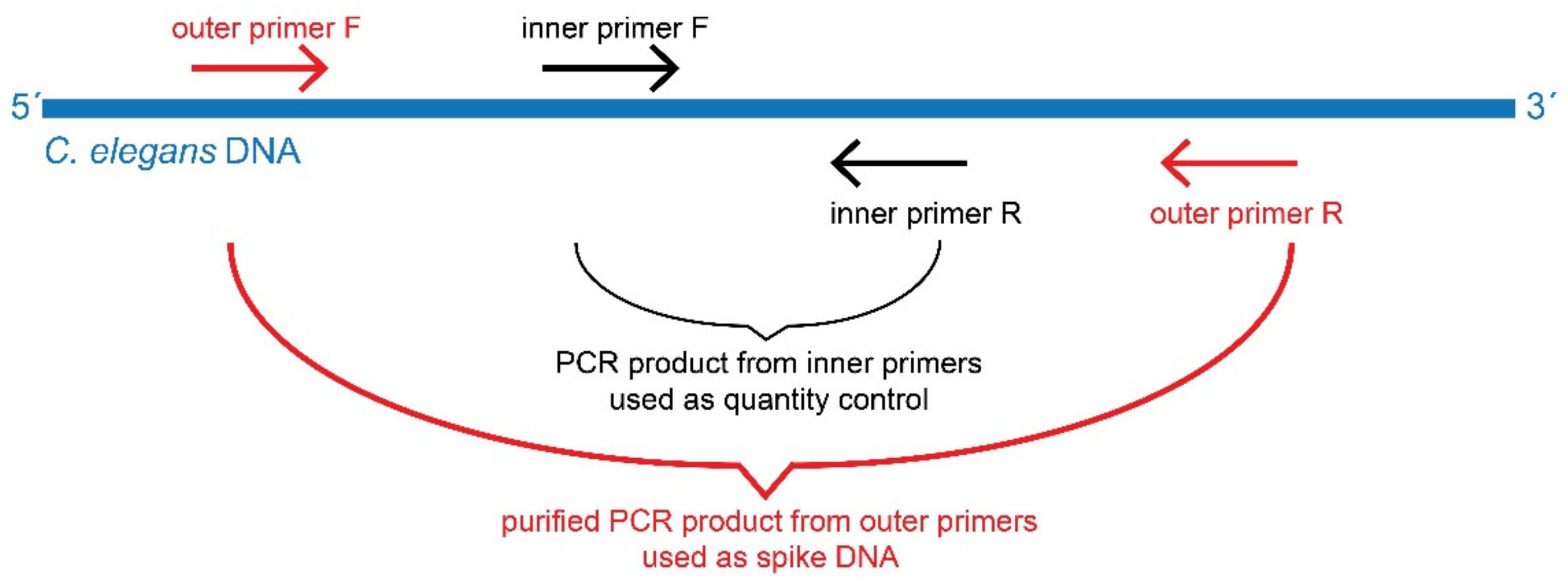
2.4. Preparation of Spike DNA
2.5. Design of the Degenerate Primers for but Gene and Analysis of PCR Products
2.6. qPCR
2.7. Gut Microbiome Taxonomic Analysis
2.8. Quantification of Butyrate in Fecal Samples by NMR Spectroscopy
2.9. Statistical Evaluation
3. Results
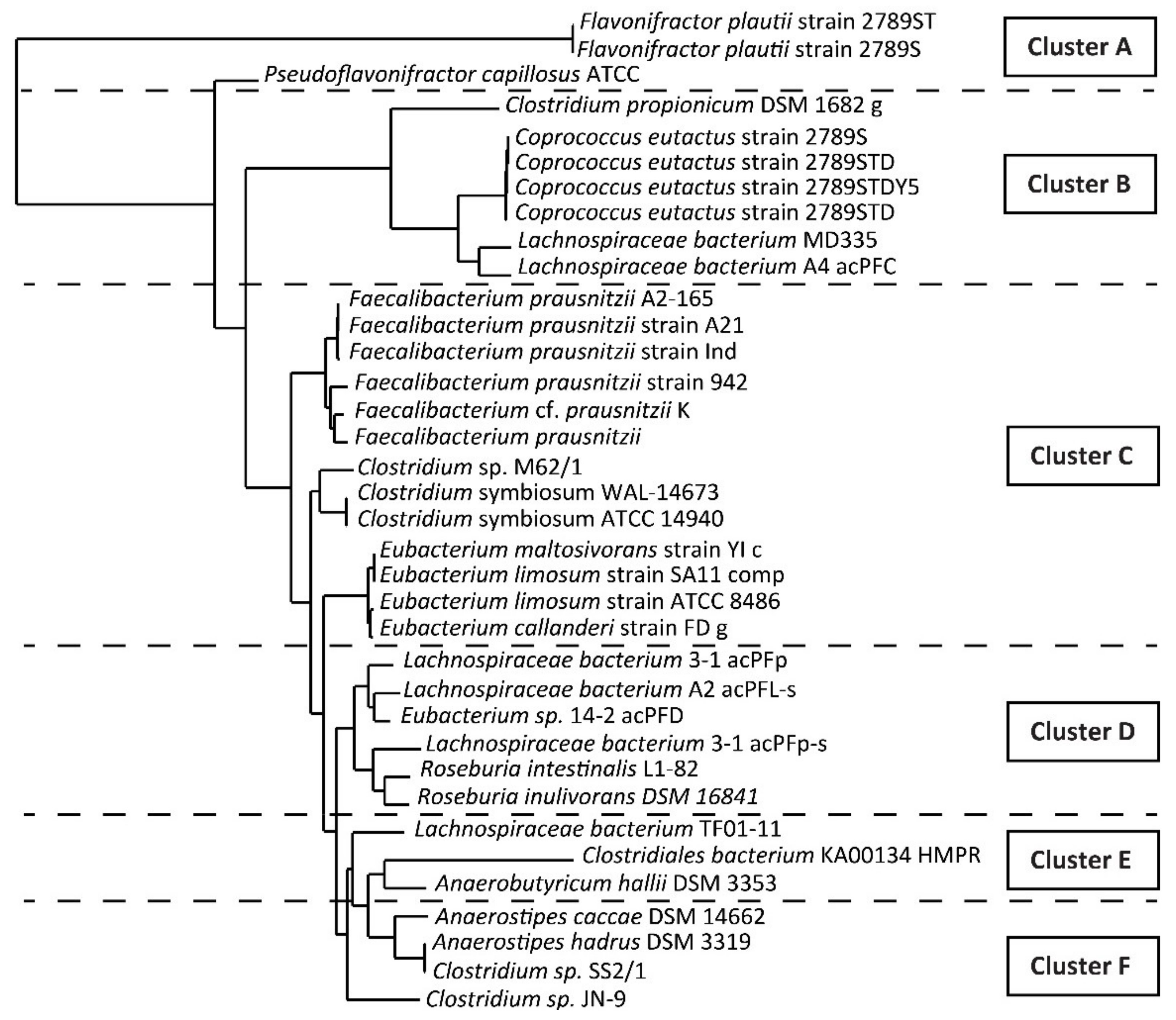
3.1. Identification of the Target Bacteria
3.2. Design of the Degenerate Primers for but Gene
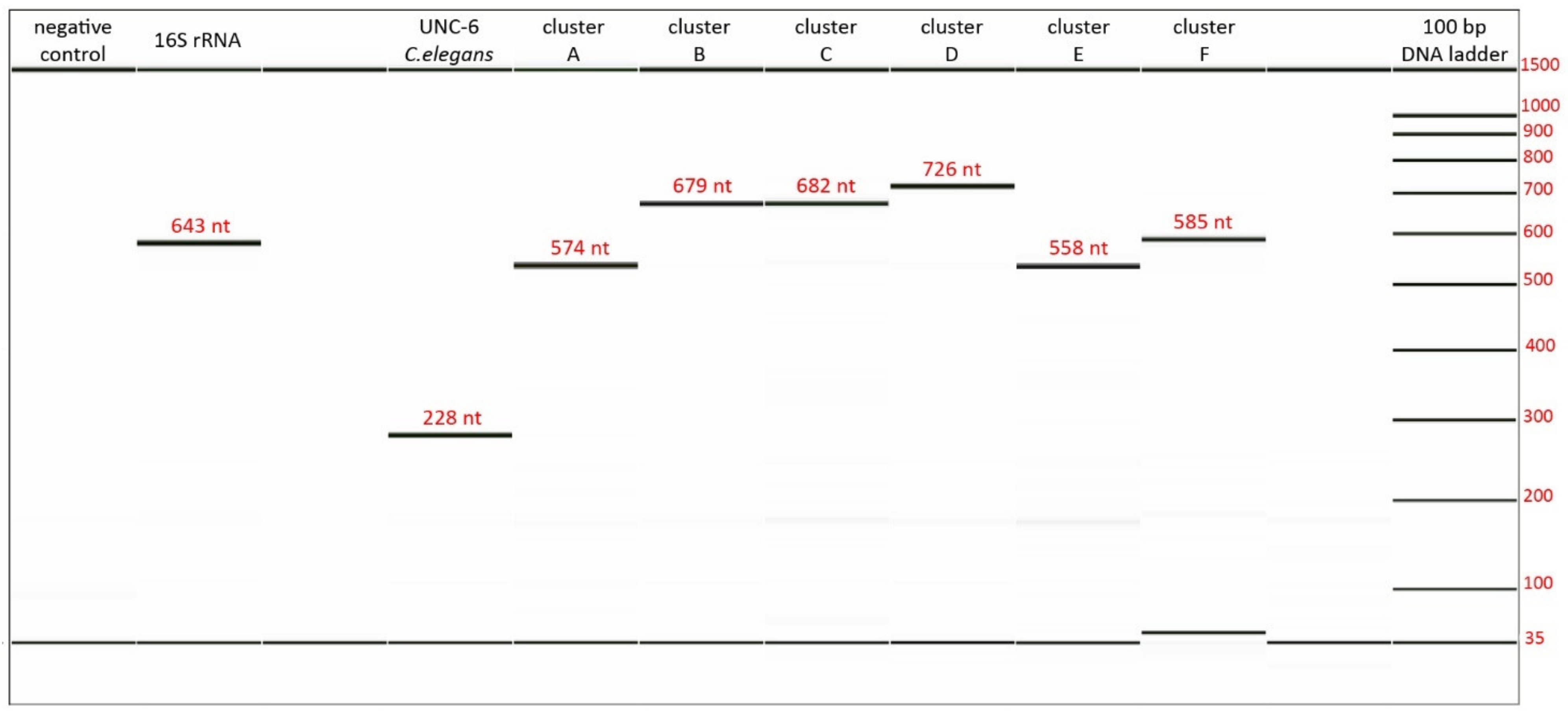
3.3. Validation and Optimization of Designed Primers
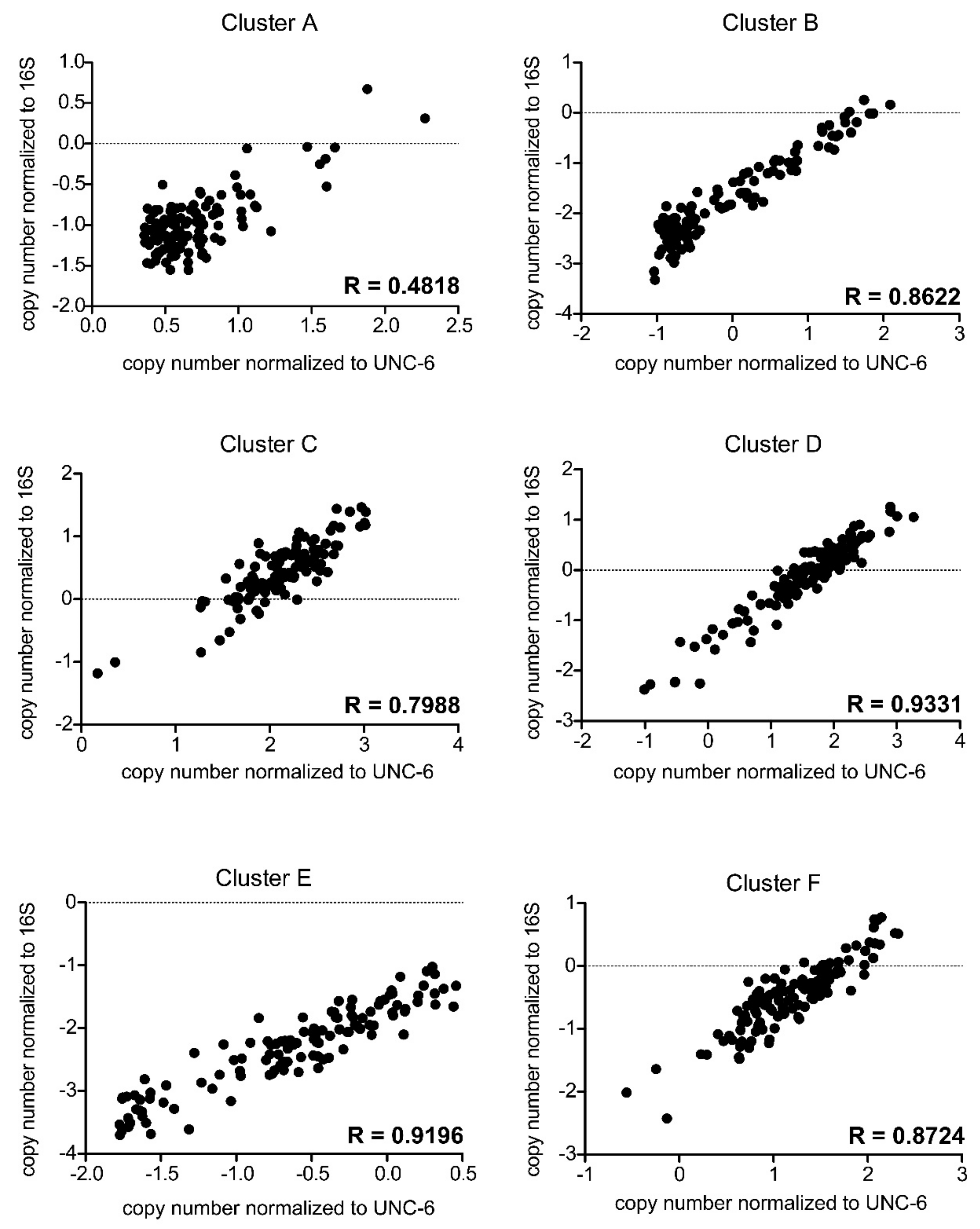
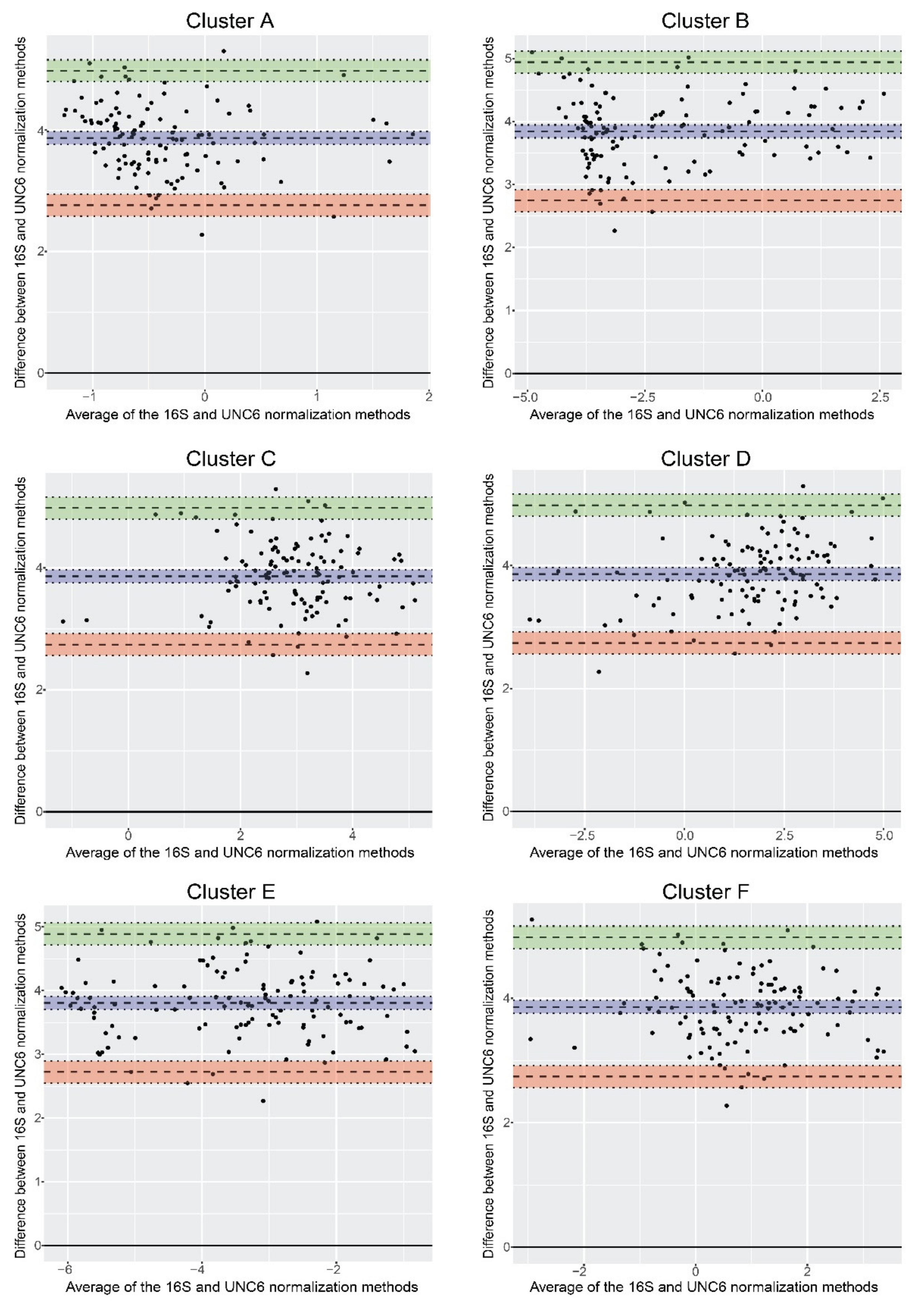
3.4. Normalization of qPCR Results
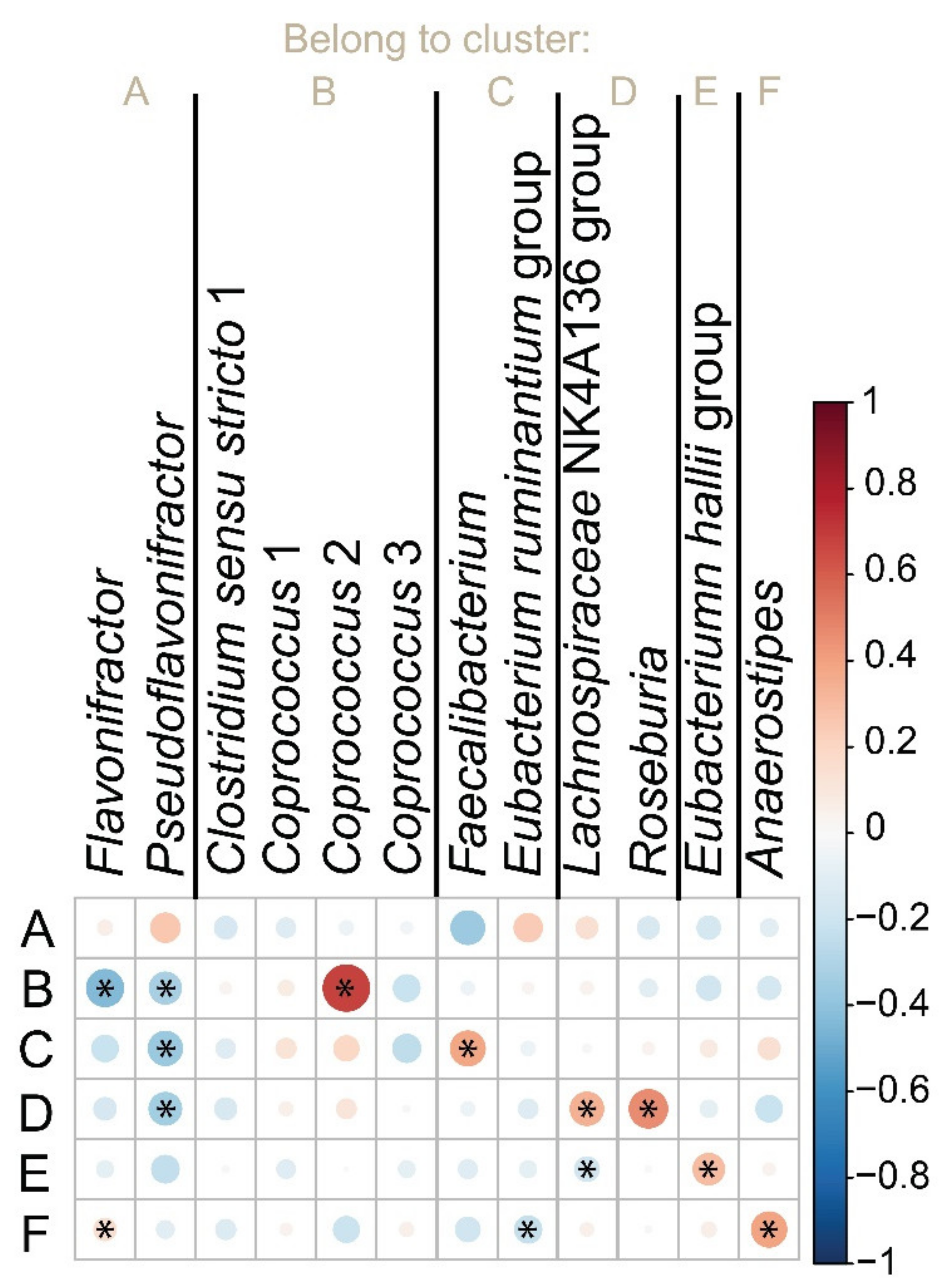
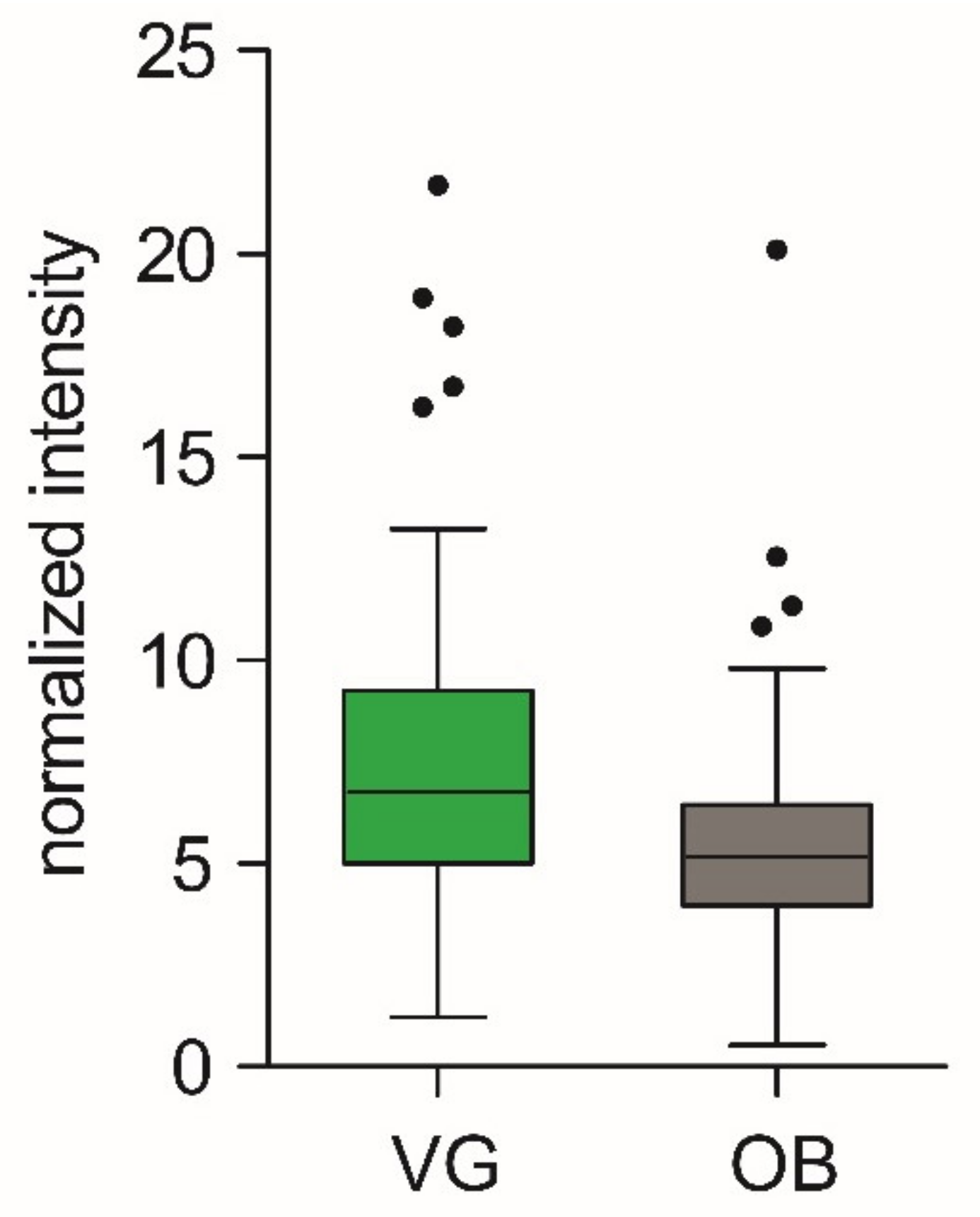
3.5. Quantification of but Gene in Populations with Contrast Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tilg, H.; Moschen, A.R. Microbiota and diabetes: An evolving relationship. Gut 2014, 63, 1513–1521. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.N.; Macia, L.; Mackay, C.R. Diet, Metabolites, and “Western-Lifestyle” Inflammatory Diseases. Immunity 2014, 40, 833–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Verbeke, K.A.; Boobis, A.R.; Chiodini, A.; Edwards, C.A.; Franck, A.; Kleerebezem, M.; Nauta, A.; Raes, J.; Van Tol, E.A.F.; Tuohy, K.M. Towards microbial fermentation metabolites as markers for health benefits of prebiotics. Nutr. Res. Rev. 2015, 28, 42–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Hernández, M.A.G.; Canfora, E.E.; Jocken, J.W.E.; Blaak, E.E. The short-chain fatty acid acetate in body weight control and insulin sensitivity. Nutrients 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Raka, F.; Adeli, K. The Role of the Gut Microbiota in Lipid and Lipoprotein Metabolism. J. Clin. Med. 2019, 8, 2227. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Lupton, J.R. Diet Induced Changes in the Colonic Environment and Colorectal Cancer. J. Nutr. 2004, 134, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Guilloteau, P.; Martin, L.; Eeckhaut, V.; Ducatelle, R.; Zabielski, R.; Van Immerseel, F. From the gut to the peripheral tissues: The multiple effects of butyrate. Nutr. Res. Rev. 2010, 23, 366–384. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, J.L.; Yap, Y.A.; McLeod, K.H.; MacKay, C.R.; Marinõ, E. Dietary metabolites and the gut microbiota: An alternative approach to control inflammatory and autoimmune diseases. Clin. Transl. Immunol. 2016, 5, e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappenden, K.A.; McBurney, M.I. Systemic short-chain fatty acids rapidly alter gastrointestinal structure, function, and expression of early response genes. Dig. Dis. Sci. 1998, 43, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Burger-van Paassen, N.; Vincent, A.; Puiman, P.J.; van der Sluis, M.; Bouma, J.; Boehm, G.; van Goudoever, J.B.; Van Seuningen, I.; Renes, I.B. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: Implications for epithelial protection. Biochem. J. 2009, 420, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef] [PubMed]
- Lampe, J.W.; Navarro, S.L.; Hullar, M.A.J.; Shojaie, A. Inter-individual differences in response to dietary intervention: Integrating omics platforms towards personalised dietary recommendations. Proc. Nutr. Soc. 2013, 72, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. Omics J. Integr. Biol. 2018, 22, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Sanschagrin, S.; Yergeau, E. Next-generation sequencing of 16S ribosomal RNA gene amplicons. J. Vis. Exp. 2014, 90, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, N.; Lipman, D.J.; Lu, Z. Towards PubMed 2.0. Elife 2017, 6, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Penton, C.R.; Wang, Q.; Young, V.B.; Antonopoulos, D.A.; Sogin, M.L.; Morrison, H.G.; Raffals, L.; Chang, E.B.; Huffnagle, G.B.; et al. A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome 2013, 1, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, S.; Kaur, H.; Mande, S.S. Comparative in silico analysis of butyrate production pathways in gut commensals and pathogens. Front. Microbiol. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Duncan, S.H.; McCrae, S.I.; Millar, J.; Jackson, M.S.; Flint, H.J. Restricted Distribution of the Butyrate Kinase Pathway among Butyrate-Producing Bacteria from the Human Colon. J. Bacteriol. 2004, 186, 2099–2106. [Google Scholar] [CrossRef] [Green Version]
- Lissemore, J.L.; Lackner, L.L.; Fedoriw, G.D.; De Stasio, E.A. Isolation of Caenorhabditis elegans genomic DNA and detection of deletions in the unc-93 gene using PCR. Biochem. Mol. Biol. Educ. 2005, 33, 219–226. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, 1–12. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, 20–25. [Google Scholar] [CrossRef]
- Apprill, A.; Mcnally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Budinska, E.; Gojda, J.; Heczkova, M.; Bratova, M.; Dankova, H.; Wohl, P.; Bastova, H.; Lanska, V.; Kostovcik, M.; Dastych, M.; et al. Microbiome and Metabolome Profiles Associated With Different Types of Short Bowel Syndrome: Implications for Treatment. J. Parenter. Enter. Nutr. 2020, 44, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Correspondence QIIME allows analysis of high- throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat. Publ. Gr. 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Pelantová, H.; Bugáňová, M.; Anýž, J.; Železná, B.; Maletínská, L.; Novák, D.; Haluzík, M.; Kuzma, M. Strategy for NMR metabolomic analysis of urine in mouse models of obesity- from sample collection to interpretation of acquired data. J. Pharm. Biomed. Anal. 2015, 115, 225–235. [Google Scholar] [CrossRef] [PubMed]
- R core Team. A language and environment for statistical computing. Found. Stat. Comput. 2013, 2, 1–12. [Google Scholar]
- Aitchison, J. The Statistical Analysis of Compositional Data. Stat. Anal. Compos. Data 1986, 44, 139–177. [Google Scholar] [CrossRef]
- Fish, J.A.; Chai, B.; Wang, Q.; Sun, Y.; Brown, C.T.; Tiedje, J.M.; Cole, J.R. FunGene: The functional gene pipeline and repository. Front. Microbiol. 2013, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; McCrae, S.I.; Charrier, C.; Flint, H.J. Organization of butyrate synthetic genes in human colonic bacteria: Phylogenetic conservation and horizontal gene transfer. FEMS Microbiol. Lett. 2007, 269, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 1–22. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, 465–469. [Google Scholar] [CrossRef]
- Lane, C.E.; Hulgan, D.; O’Quinn, K.; Benton, M.G. CEMAsuite: Open source degenerate PCR primer design. Bioinformatics 2015, 31, 3688–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Leigh Greathouse, K.; Sinha, R.; Vogtmann, E. DNA extraction for human microbiome studies: The issue of standardization. BMC Microbiol. 2018, 18, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, S.V.; Vang, K.B.; Gies, A.; Griffin, R.J.; Jun, S.; Nookaew, I.; Dings, R.P.M. Sample storage conditions induce post- collection biases in microbiome profiles. BMC Microbiol. 2018, 18, 227. [Google Scholar] [CrossRef] [PubMed]
- Větrovský, T.; Baldrian, P. The Variability of the 16S rRNA Gene in Bacterial Genomes and Its Consequences for Bacterial Community Analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinu, M.; Abbate, R.; Gensini, G.F.; Casini, A.; Sofi, F. Vegetarian, vegan diets and multiple health outcomes: A systematic review with meta-analysis of observational studies. Crit. Rev. Food Sci. Nutr. 2017, 57, 3640–3649. [Google Scholar] [CrossRef]
- Jia, W.; Zhen, J.; Liu, A.; Yuan, J.; Wu, X.; Zhao, P.; Zhao, L.; Li, X.; Liu, Q.; Huang, G.; et al. Long-Term Vegan Meditation Improved Human Gut Microbiota. Evid. Based Complement. Altern. Med. 2020, 2020, 9517897. [Google Scholar] [CrossRef]
- Wu, G.D.; Compher, C.; Chen, E.Z.; Smith, S.A.; Shah, R.D.; Bittinger, K.; Chehoud, C.; Albenberg, L.G.; Nessel, L.; Gilroy, E.; et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut 2016, 65, 63–72. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′ to 3′) |
|---|---|
| inner primer forward | GAAGAGCAAGATCAGTGTTC |
| inner primer reverse | CTTGCAAATGACACCTTG |
| outer primer forward | GTAATCGTTGTGCCAAAGG |
| outer primer reverse | TCCTCCCATTCCACCAATAC |
| Taxonomy (including the Strain) | Nucleotide Sequence Accession Number | Protein Sequence Accession Number |
|---|---|---|
| Anaerostipes caccae DSM 14662 | ABAX03000012 | WP_006566634 |
| Anaerobutyricum hallii DSM 3353 | ACEP01000025 | EEG37758 |
| Anaerostipes hadrus DSM 3319 | AMEY01000089 | EKY19441 |
| Clostridiales bacterium KA00134 | LTAF01000006 | KXO16903 |
| Clostridium sp. JN-9 | CP035280 | QAT39812 |
| Clostridium sp. M62/1 | ACFX02000051 | EFE10856 |
| Clostridium sp. SS2/1 | ABGC03000034 | EDS21983 |
| [Clostridium] propionicum DSM 1682 | FQUA01000004 | SHE65336 |
| [Clostridium] symbiosum ATCC 14940 | AWSU01000039 | ERI80067 |
| [Clostridium] symbiosum WAL-14673 | ADLR01000107 | EGB17928 |
| Coprococcus eutactus 2789STDY5608829 | CYYZ01000002 | CUN77211 |
| Coprococcus eutactus 2789STDY5608843 | CYYJ01000005 | CUO17024 |
| Coprococcus eutactus 2789STDY5608888 | CYYE01000001 | CUN69525 |
| Coprococcus eutactus 2789STDY5834963 | CYXU01000007 | CUN05838 |
| Eubacterium callanderi FD | FRBP01000012 | SHM18802 |
| Eubacterium limosum ATCC 8486 | CP019962 | ARD67787 |
| Eubacterium limosum SA11 | CP011914 | ALU15403 |
| Eubacterium maltosivorans YI | CP029487 | QCT73558 |
| Eubacterium sp. 14-2 | ASSS01000012 | EOT23498 |
| Faecalibacterium prausnitzii 942/30-2 | CP026548 | AXA81262 |
| Faecalibacterium prausnitzii A2165 CG447_00005 | CP022479 | ATO98751 |
| Faecalibacterium prausnitzii A2-165 FAEPRAA2165_01562 | ACOP02000044 | EEU96797 |
| Faecalibacterium prausnitzii APC918/95b | CP030777 | AXB28579 |
| Faecalibacterium prausnitzii Indica | CP023819 | ATL89114 |
| Faecalibacterium prausnitzii KLE1255 | AECU01000083 | EFQ07628 |
| Flavonifractor plautii 2789STDY5834892 | CZAS01000006 | CUP57950 |
| Flavonifractor plautii 2789STDY5834932 | CZBD01000023 | CUQ37563 |
| Lachnospiraceae bacterium 3-1 | ASST01000018 | EOS23550 |
| Lachnospiraceae bacterium 3-1 | ASST01000032 | EOS21051 |
| Lachnospiraceae bacterium A2 | ASSX01000004 | EOS48506 |
| Lachnospiraceae bacterium A4 | ASSR01000007 | EOS36856 |
| Lachnospiraceae bacterium MD335 | ASSW01000016 | EOS51721 |
| Lachnospiraceae bacterium TF01-11 | LLKB01000001 | KQC86641 |
| Pseudoflavonifractor capillosus ATCC 29799 | AAXG02000004 | EDN01706 |
| Roseburia intestinalis L1-82 | ABYJ02000099 | EEV00989 |
| Roseburia inulinivorans DSM 16841 | ACFY01000152 | EEG92587 |
| Primer Name | Primer Sequence (5′ to 3′) | Expected Product Length (nt) |
|---|---|---|
| but cluster A forward | MCTGGGYATYCACACCGAG | 574 |
| but cluster A reverse | GGTGGGCGATGGAGATAA | |
| but cluster B forward | GGKCCBATHGARRTTGCAGA | 679 |
| but cluster B reverse | TKTCGTCMASCCABTCATAC | |
| but cluster C forward | GBGACTGGSTRGATTAYG | 682 |
| but cluster C reverse | TCVACRTACATYTCSGTGTG | |
| but cluster D forward | TGGAAYTCMTGGCATATGTC | 726 |
| but cluster D reverse | VGMRTTGTTRATGGAMATAAA | |
| but cluster E forward | TGHAGSABHTSWTTTTACATGGA | 558 |
| but cluster E reverse | SSCTTTGCAATGTCAACAAA | |
| but cluster F forward | AAATATGCCTCGHTGCYTWG | 585 |
| but cluster F reverse | ARRTARGCACCYAWAACGAAATC |
| Cluster A | Cluster B | Cluster C | Cluster D | Cluster E | Cluster F | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UNC | 16S | UNC | 16S | UNC | 16S | UNC | 16S | UNC | 16S | UNC | 16S | |
| VG | 3.5 (2.0) | 0.08 (0.06) | 0.45 (5.09) | 0.01 (0.09) | 211 (243) | 4.9 (5.8) | 63 (118) | 1.1 (2.4) | 0.28 (0.48) | 0.01 (0.01) | 12.5 (25.1) | 0.34 (0.41) |
| OB | 4.8 (3.5) | 0.10 (0.10) | 0.28 (1.91) | 0.01 (0.05) | 86 (71) | 1.8 (1.6) | 34 (76) | 0.9 (1.6) | 0.32 (0.61) | 0.01 (0.01) | 17.6 (30.9) | 0.38 (0.70) |
| p-value | 0.004 | 0.019 | 0.942 | 0.438 | <0.001 | <0.001 | 0.167 | 0.225 | 0.769 | 0.840 | 0.680 | 0.589 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daskova, N.; Heczkova, M.; Modos, I.; Videnska, P.; Splichalova, P.; Pelantova, H.; Kuzma, M.; Gojda, J.; Cahova, M. Determination of Butyrate Synthesis Capacity in Gut Microbiota: Quantification of but Gene Abundance by qPCR in Fecal Samples. Biomolecules 2021, 11, 1303. https://doi.org/10.3390/biom11091303
Daskova N, Heczkova M, Modos I, Videnska P, Splichalova P, Pelantova H, Kuzma M, Gojda J, Cahova M. Determination of Butyrate Synthesis Capacity in Gut Microbiota: Quantification of but Gene Abundance by qPCR in Fecal Samples. Biomolecules. 2021; 11(9):1303. https://doi.org/10.3390/biom11091303
Chicago/Turabian StyleDaskova, Nikola, Marie Heczkova, Istvan Modos, Petra Videnska, Petra Splichalova, Helena Pelantova, Marek Kuzma, Jan Gojda, and Monika Cahova. 2021. "Determination of Butyrate Synthesis Capacity in Gut Microbiota: Quantification of but Gene Abundance by qPCR in Fecal Samples" Biomolecules 11, no. 9: 1303. https://doi.org/10.3390/biom11091303
APA StyleDaskova, N., Heczkova, M., Modos, I., Videnska, P., Splichalova, P., Pelantova, H., Kuzma, M., Gojda, J., & Cahova, M. (2021). Determination of Butyrate Synthesis Capacity in Gut Microbiota: Quantification of but Gene Abundance by qPCR in Fecal Samples. Biomolecules, 11(9), 1303. https://doi.org/10.3390/biom11091303