
HMGA1, Moonlighting Protein Function, and Cellular Real Estate: Location, Location, Location!

Abstract
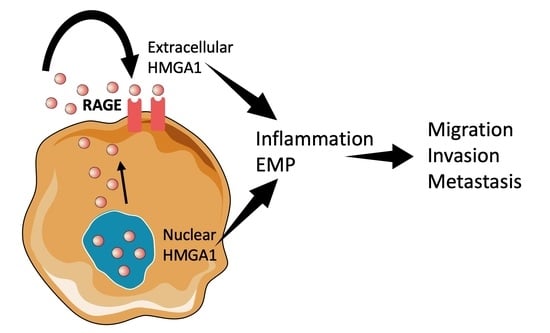
:
{kind=link}
{kind=link}
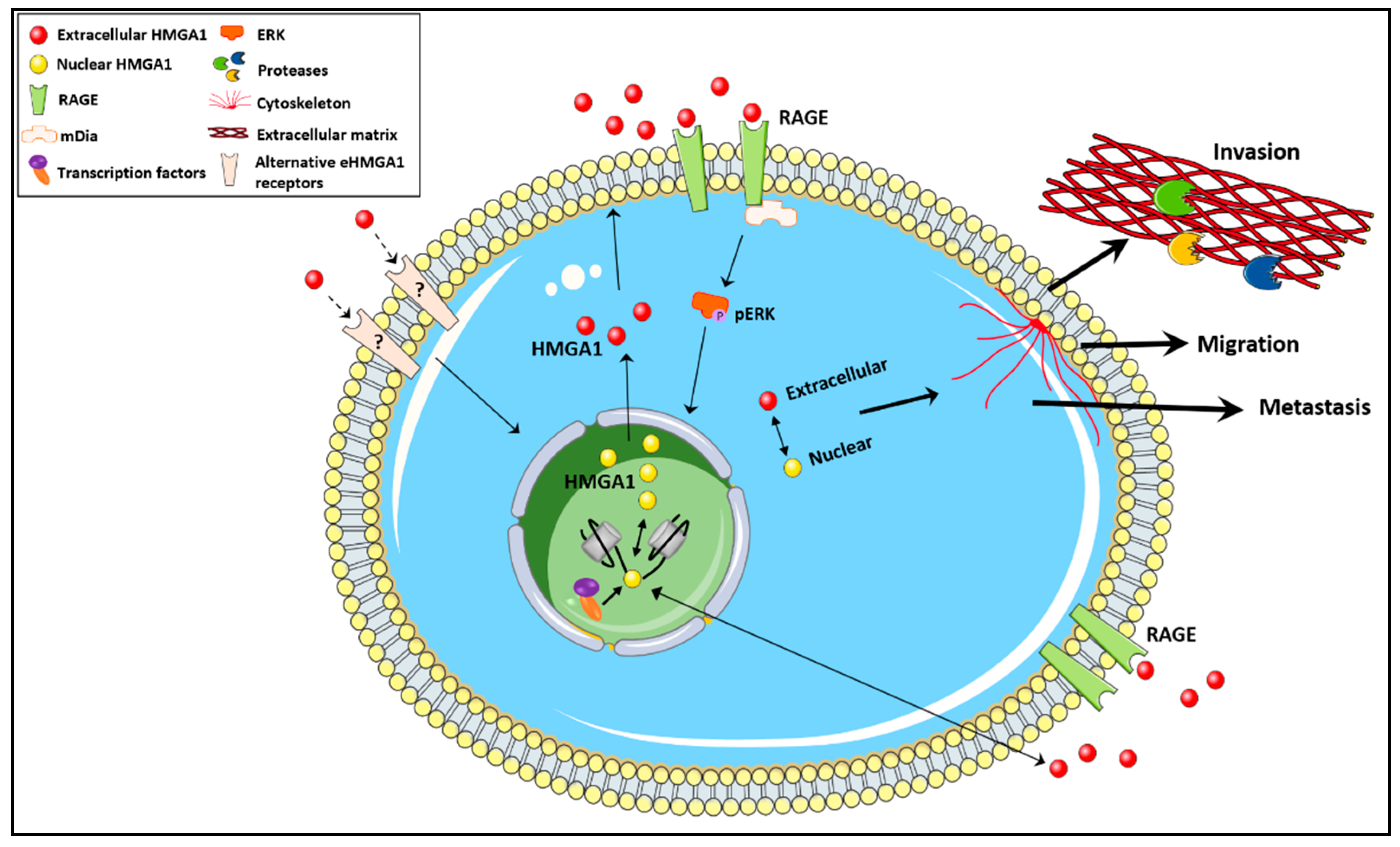{kind=link}
1. Introduction
2. Unconventional Protein Secretion (UPS)
2.1. Unconventional Secretion: UPS Pathways (I–IV)
2.2. Role of Unconventional Secretion in Cancer Biology
3. High Mobility Group Proteins and Cancer
4. Extracellular Oncogenic Role of HMGA1
4.1. HMGA1 Secretion and Casein Kinase 2 (CK2)
4.2. Receptor for Advanced Glycation End Products (RAGE)
5. Reconciling HMGA1 Functions across Compartments
5.1. Integrating the Inflammatory Response
5.2. Epithelial-to-Mesenchymal Plasticity (EMP)
6. Conclusions and Future Directions
- (1)
- What factors dictate the location of HMGA1?
- (2)
- Are posttranslational modifications involved (similar to those described for HMGB1)?
- (3)
- What triggers secretion together with CK2 or independent of CK2?
- (4)
- Which UPS pathway leads to HMGA1 secretion?
- (5)
- Do receptors other than RAGE mediate eHMGA1 function?
- (6)
- How does intranuclear HMGA1 collaborate with eHMGA1?
- (7)
- Does eHMGA1 function in embryonic development
- (8)
- Is HMGA1 secreted in vivo?
- (9)
- Do diverse tumors secrete HMGA1?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smalheiser, N.R. Proteins in unexpected locations. Mol. Biol. Cell 1996, 7, 1003–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, G.S.; Overall, C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009, 8, 935–948. [Google Scholar] [CrossRef]
- Jobin, X.P.G.; Solis, X.N.; Machado, Y.; Bell, X.P.A.; Rai, S.K.; Kwon, N.H.; Kim, S.; Overall, C.M.; Butler, G.S. Moonlighting matrix metalloproteinase substrates: Enhancement of proinflammatory functions of extracellular tyrosyl-tRNA synthetase upon cleavage. J. Biol. Chem. 2020, 295, 2186–2202. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-cantú, A.; Ascencio, D.; Herrera-basurto, S.; Xu, J.; Roguev, A. Protein Moonlighting Revealed by Noncatalytic Phenotypes of Yeast Enzymes. Genetics 2018, 208, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Arnoys, E.J.; Wang, J.L. Dual localization: Proteins in extracellular and intracellular compartments. Acta Histochem. 2007, 109, 89–110. [Google Scholar] [CrossRef]
- Eustace, B.K.; Sakurai, T.; Stewart, J.K.; Yimlamai, D.; Unger, C.; Zehetmeier, C.; Lain, B.; Torella, C.; Henning, S.W.; Beste, G.; et al. Functional proteomic screens reveal an essential extracellular role for hsp90α in cancer cell invasiveness. Nat. Cell Biol. 2004, 6, 507–514. [Google Scholar] [CrossRef]
- Greenberg, Y.; King, M.; Kiosses, W.; Ewalt, K.; Yang, X.; Schimmel, P.; Reader, J.; Tzima, E. The novel fragment of tyrosyl tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J. 2008, 22, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Kim, H.; Min, Y.; Choi, E.; Shin, Y.; Park, B.; Lee, S.; Kim, S. Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. USA 2005, 102, 6356–6361. [Google Scholar] [CrossRef] [Green Version]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez, O.; Peg, V.; Pujals, M.; Fern, Y.; Matres, A.; Valeri, M.; Gregori, J.; Villarreal, L.; Arribas, J.; Villanueva, J. Extracellular HMGA1 Promotes Tumor Invasion and Metastasis in Triple-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 6367–6382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, A.; Rapoport, T.; van den Berg, B. Protein translocation by the Sec61/SecY channel. Annu. Rev. Cell Dev. Biol. 2005, 21, 529–550. [Google Scholar] [CrossRef]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef] [Green Version]
- Nickel, W.; Seedorf, M. Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu. Rev. Cell Dev. Biol. 2008, 24, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Zehe, C.; Engling, A.; Wegehingel, S.; Schäfer, T.; Nickel, W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc. Natl. Acad. Sci. USA 2006, 103, 15479–15484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, W. The unconventional secretory machinery of fibroblast growth factor 2. Traffic 2011, 12, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, T.; Zentgraf, H.; Zehe, C.; Brügger, B.; Bernhagen, J.; Nickel, W. Unconventional Secretion of Fibroblast Growth Factor 2 Is Mediated by Direct Translocation across the Plasma Membrane of Mammalian Cells. J. Biol. Chem. 2004, 279, 6244–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backhaus, R.; Zehe, C.; Wegehingel, S.; Kehlenbach, A.; Schwappach, B.; Nickel, W. Unconventional protein secretion: Membrane translocation of FGF-2 does not require protein unfolding. J. Cell Sci. 2004, 117, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Christensen, P.U.; Davey, J.; Nielsen, O. The Schizosaccharomyces pombe mam1 gene encodes an ABC transporter mediating secretion of M-factor. Mol. Gen. Genet. MGG 1997, 255, 226–236. [Google Scholar] [CrossRef]
- Cabral, M.; Anjard, C.; Malhotra, V.; Loomis, W.F.; Kuspa, A. Unconventional secretion of AcbA in Dictyostelium discoideum through a vesicular intermediate. Eukaryot. Cell 2010, 9, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Duran, J.; Anjard, C.; Stefan, C.; Loomis, W.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Manjithaya, R.; Anjard, C.; Loomis, W.F.; Subramani, S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010, 188, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Stahl, P.D.; Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Souza-schorey, C.D.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.S.; Moyer, B.D.; Bannykh, S.; Yoo, H.M.; Riordan, J.R.; Balch, W.E. Non-conventional Trafficking of the Cystic Fibrosis Transmembrane Conductance Regulator through the Early Secretory Pathway. J. Biol. Chem. 2002, 277, 11401–11409. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Garcia, D.; Brouwers, N.; Duran, J.M.; Mora, G.; Curwin, A.J.; Malhotra, V. A diacidic motif determines unconventional secretion of wild-type and ALS-linked mutant SOD1. J. Cell Biol. 2017, 216, 2691–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, F.; Grieve, A.; Rabouille, C. Unconventional secretion: A stress on GRASP. Curr. Opin. Cell Biol. 2011, 23, 498–504. [Google Scholar] [CrossRef]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2008, 10, 148–155. [Google Scholar] [CrossRef]
- Ogura, Y.; Sutterwala, F.S.; Flavell, R.A. The Inflammasome: First Line of the Immune Response to Cell Stress. Cell 2006, 126, 659–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardaway, A.L.; Podgorski, I. IL-1β, RAGE and FABP4: Targeting the dynamic trio in metabolic inflammation and related pathologies. Future Med. Chem. 2013, 5, 1089–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; You, H.; Li, X.; Liu, T.; Wang, P.; Wang, B. HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by activation of p38 MAPK and NF-κB through receptors for advanced glycation end-products in macrophages. Asian Pac. J. Cancer Prev. 2012, 13, 1365–1370. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Ru, A.; Werner, S.; Beer, H. Active Caspase-1 Is a Regulator of Unconventional Protein Secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Jiyoon, K.; Heon Yung, G.; Min Goo, L. Unconventional protein secretion – new insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef] [Green Version]
- Mendez, O.; Pérez, J.; Soberino, J.; Racca, F.; Cortes, J.; Villanueva, J. Clinical Implications of Extracellular HMGA1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and Basic Amino Acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R. Nuclear functions of the HMG proteins. Biochim. Biophys. Acta - Gene Regul. Mech. 2010, 1799, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlitz, G.; Hock, R.; Ueda, T.; Bustin, M. The dynamics of HMG protein-chromatin interactions in living cells. Biochem. Cell Biol. 2009, 87, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Kwak, M.S.; Lee, B.; Shin, J.M.; Aum, S.; Park, I.H.; Lee, M.G.; Shin, J.S. Secretory autophagy machinery and vesicular trafficking are involved in HMGB1 secretion. Autophagy 2020. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; Frankel, A.E.; Thorburn, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009, 16, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Postnikov, Y.; Bustin, M. Regulation of chromatin structure and function By HMGN proteins. Biochim. Biophys. Acta - Gene Regul. Mech. 2010, 1799, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Catez, F.; Brown, D.T.; Misteli, T.; Bustin, M. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep. 2002, 3, 760–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; West, K.L.; Rubinstein, Y.; Bergel, M.; Postnikov, Y.V.; Bustin, M. Chromosomal protein HMGN1 enhances the acetylation of lysine 14 in histone H3. EMBO J. 2005, 24, 3038–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Tewary, P.; de la Rosa, G.; Wei, F.; Oppenheim, J.J. The alarmin functions of high-mobility group proteins. Physiol. Behav. 2010, 1799, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Park, I.A.; Heo, S.-H.; Song, I.H.; Kim, Y.-A.; Park, H.S.; Bang, W.S.; Park, S.Y.; Jo, J.-H.; Lee, H.J.; Gong, G. Endoplasmic reticulum stress induces secretion of high-mobility group proteins and is associated with tumor-infiltrating lymphocytes in triple-negative breast cancer. Oncotarget 2016, 7, 59957–59964. [Google Scholar] [CrossRef] [Green Version]
- Resar, L.M.S. The High Mobility Group A1 Gene: Transforming Inflammatory Signals into Cancer? Cancer Res. 2010, 70, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef]
- Banks, G.C.; Mohr, B.; Reeves, R. The HMG-I(Y) A · T-hook peptide motif confers DNA-binding specificity to a structured chimeric protein. J. Biol. Chem. 1999, 274, 16536–16544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geierstanger, B.H.; Volkman, B.F.; Kremer, W.; Wemmer, D.E. Short Peptide Fragments Derived from HMG-I/Y Proteins Bind Specifically to the Minor Groove of DNA. Biochemistry 1994, 33, 5347–5355. [Google Scholar] [CrossRef]
- Chiappetta, G.; Avantaggiato, V.; Visconti, R.; Fedele, M.; Battista, S.; Trapasso, F.; Merciai, B.M.; Fidanza, V.; Giancotti, V.; Santoro, M.; et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996, 13, 2439–2446. [Google Scholar] [PubMed]
- Shah, S.N.; Kerr, C.; Cope, L.; Zambidis, E.; Liu, C.; Hillion, J.; Belton, A.; Huso, D.L.; Resar, L.M.S. HMGA1 Reprograms Somatic Cells into Pluripotent Stem Cells by Inducing Stem Cell Transcriptional Networks. PLoS ONE 2012, 7, e48533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristov, A.C.; Cope, L.; Reyes, M.D.; Singh, M.; Iacobuzio-Donahue, C.; Maitra, A.; Resar, L.M.S. HMGA2 protein expression correlates with lymph node metastasis and increased tumor grade in pancreatic ductal adenocarcinoma. Mod. Pathol. 2009, 22, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Di Cello, F.; Kowalski, J.; Hristov, A.C.; Tsai, H.L.; Bhojwani, D.; Meyer, J.A.; Carroll, W.L.; Belton, A.; Resar, L.M.S. HMGA1 overexpression correlates with relapse in childhood B-lineage acute lymphoblastic leukemia. Leuk. Lymphoma 2013, 54, 2565–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristov, A.C.; Cope, L.; Di Cello, F.; Reyes, M.D.; Singh, M.; Hillion, J.A.; Belton, A.; Joseph, B.; Schuldenfrei, A.; Iacobuzio-Donahue, C.A.; et al. HMGA1 correlates with advanced tumor grade and decreased survival in pancreatic ductal adenocarcinoma. Mod. Pathol. 2010, 23, 98–104. [Google Scholar] [CrossRef]
- Gorbounov, M.; Carleton, N.M.; Asch-Kendrick, R.J.; Xian, L.; Rooper, L.; Chia, L.; Cimino-Mathews, A.; Cope, L.; Meeker, A.; Stearns, V.; et al. High mobility group A1 (HMGA1) protein and gene expression correlate with ER-negativity and poor outcomes in breast cancer. Breast Cancer Res. Treat. 2020, 179, 25–35. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Xu, Y.; Sumter, T.F.; Bhattacharya, R.; Tesfaye, A.; Fuchs, E.J.; Wood, L.J.; Huso, D.L.; Resar, L.M.S. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004, 64, 3371–3375. [Google Scholar] [CrossRef] [Green Version]
- Schuldenfrei, A.; Belton, A.; Kowalski, J.; Talbot, C.C.; Di Cello, F.; Poh, W.; Tsai, H.L.; Shah, S.N.; Huso, T.H.; Huso, D.L.; et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics 2011, 12, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, R.; Beckerbauer, L. HMGI/Y proteins: Flexible regulators of transcription and chromatin structure. Biochim. Biophys. Acta - Gene Struct. Expr. 2001, 1519, 13–29. [Google Scholar] [CrossRef]
- Lanahan, A.; Williams, J.B.; Sanders, L.K.; Nathans, D. Growth factor-induced delayed early response genes. Mol. Cell. Biol. 1992, 12, 3919–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holth, L.T.; Thorlacius, A.E.; Reeves, R. Effects of epidermal growth factor and estrogen on the regulation of the HMG-I/Y gene in human mammary epithelial cell lines. DNA Cell Biol. 1997, 16, 1299–1309. [Google Scholar] [CrossRef]
- Cleynen, I.; Huysmans, C.; Sasazuki, T.; Shirasawa, S.; Van De Ven, W.; Peeters, K. Transcriptional control of the human high mobility group A1 gene: Basal and oncogenic Ras-regulated expression. Cancer Res. 2007, 67, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Wood, L.J.; Mukherjee, M.; Dolde, C.E.; Xu, Y.; Maher, J.F.; Bunton, T.E.; Williams, J.B.; Resar, L.M.S. HMG-I/Y, a New c-Myc Target Gene and Potential Oncogene. Mol. Cell. Biol. 2000, 20, 5490–5502. [Google Scholar] [CrossRef] [Green Version]
- Giannini, G.; Cerignoli, F.; Mellone, M.; Massimi, I.; Ambrosi, C.; Rinaldi, C.; Dominici, C.; Frati, L.; Screpanti, I.; Gulino, A. High mobility group A1 is a molecular target for MYCN in human neuroblastoma. Cancer Res. 2005, 65, 8308–8316. [Google Scholar] [CrossRef] [Green Version]
- Xian, L.; Georgess, D.; Huso, T.; Cope, L.; Belton, A.; Chang, Y.T.; Kuang, W.; Gu, Q.; Zhang, X.; Senger, S.; et al. HMGA1 amplifies Wnt signalling and expands the intestinal stem cell compartment and Paneth cell niche. Nat. Commun. 2017, 8, 15008. [Google Scholar] [CrossRef] [Green Version]
- Palvimo, J.; Linnala-Kankkunen, A. Identification of sites on chromosomal protein HMG-I phosphorylated by casein kinase II. Elsevier Sci. Publ. 1989, 257, 101–104. [Google Scholar] [CrossRef] [Green Version]
- de Abreu da Silva, I.C.; Carneiro, V.C.; de Moraes Maciel, R.; da Costa, R.F.M.; Furtado, D.R.; de Oliveira, F.M.B.; da Silva-Neto, M.A.C.; Rumjanek, F.D.; Fantappié, M.R. CK2 Phosphorylation of Schistosoma mansoni HMGB1 Protein Regulates Its Cellular Traffic and Secretion but Not Its DNA Transactions. PLoS ONE 2011, 6, e23572. [Google Scholar] [CrossRef]
- Pencheva, N.; de Gooijer, M.C.; Vis, D.J.; Wessels, L.F.A.; Würdinger, T.; van Tellingen, O.; Bernards, R. Identification of a Druggable Pathway Controlling Glioblastoma Invasiveness. Cell Rep. 2017, 20, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, B.M.; Boewe, A.S.; Götz, C.; Philipp, S.E.; Urbschat, S.; Oertel, J.; Menger, M.D.; Laschke, M.W.; Ampofo, E. Ck2 activity mediates the aggressive molecular signature of glioblastoma multiforme by inducing nerve/glial antigen (Ng)2 expression. Cancers 2021, 13, 1678. [Google Scholar] [CrossRef] [PubMed]
- Nitta, R.T.; Gholamin, S.; Feroze, A.H.; Agarwal, M.; Cheshier, S.H.; Mitra, S.S.; Li, G. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene 2015 3428 2014, 34, 3688–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pan, S.; Tsai, C.; Kuo, T.; Hsu, Y. Phosphoproteomics Reveals HMGA1, a CK2 Substrate, as a Drug-Resistant Target in Non-Small Cell Lung Cancer. Nat. Publ. Gr. 2017, 7, 44021. [Google Scholar] [CrossRef] [Green Version]
- Sessa, L.; Gatti, E.; Zeni, F.; Antonelli, A.; Catucci, A.; Koch, M.; Pompilio, G.; Fritz, G.; Raucci, A.; Bianchi, M.E. The Receptor for Advanced Glycation End-products (RAGE) is only present in mammals, and belongs to a family of Cell Adhesion Molecules (CAMs). PLoS ONE 2014, 9, e86903. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Demling, N.; Ehrhardt, C.; Kasper, M.; Laue, M.; Knels, L.; Rieber, E.P. Promotion of cell adherence and spreading: A novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006, 323, 475–488. [Google Scholar] [CrossRef]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, annurev. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Chen, M.C.; Chen, K.C.; Chang, G.C.; Lin, H.; Wu, C.C.; Kao, W.H.; Teng, C.L.J.; Hsu, S.L.; Yang, T.Y. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. 2020, 11, 265. [Google Scholar] [CrossRef]
- Chen, R.C.; Yi, P.P.; Zhou, R.R.; Xiao, M.F.; Huang, Z.B.; Tang, D.L.; Huang, Y.; Fan, X.G. The role of HMGB1-RAGE axis in migration and invasion of hepatocellular carcinoma cell lines. Mol. Cell. Biochem. 2014, 390, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Li, X.; Hua, Z.; Ma, J.; Wu, X.; Liu, Z.; Chen, H.; Cui, Z. S100A7 promotes the migration, invasion and metastasis of human cervical cancer cells through epithelial-mesenchymal transition. Oncotarget 2017, 8, 24964–24977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Body-Malapel, M.; Djouina, M.; Waxin, C.; Langlois, A.; Gower-Rousseau, C.; Zerbib, P.; Schmidt, A.M.; Desreumaux, P.; Boulanger, E.; Vignal, C. The RAGE signaling pathway is involved in intestinal inflammation and represents a promising therapeutic target for Inflammatory Bowel Diseases. Mucosal Immunol. 2019, 12, 468–478. [Google Scholar] [CrossRef]
- Resar, L.; Hillion, J.; Dhara, S.; Sumter, T.F.; Mukherjee, M.; Di Cello, F.; Belton, A.; Turkson, J.; Jaganathan, S.; Cheng, L.; et al. The HMGA1a-STAT3 axis: An “Achilles Heel” for Hematopoietic Malignancies Overexpressing HMGA1a? Blood 2008, 112, 3810. [Google Scholar] [CrossRef]
- Hillion, J.; Belton, A.M.; Shah, S.N.; Turkson, J.; Jing, N.; Tweardy, D.J.; Di Cello, F.; Huso, D.L.; Resar, L.M.S. Nanoparticle delivery of inhibitory signal transducer and activator of transcription 3 G-quartet oligonucleotides blocks tumor growth in HMGA1 transgenic model of T-cell leukemia. Leuk. Lymphoma 2014, 55, 1194–1197. [Google Scholar] [CrossRef] [Green Version]
- Arcaini, L.; Cazzola, M. Benefits and risks of JAK inhibition. Blood 2018, 132, 675–676. [Google Scholar] [CrossRef] [Green Version]
- Di Cello, F.; Hillion, J.; Kowalski, J.; Ronnett, B.M.; Aderinto, A.; Huso, D.L.; Resar, L.M.S. Cyclooxygenase inhibitors block uterine tumorigenesis in HMGA1a transgenic mice and human xenografts. Mol. Cancer Ther. 2008, 7, 2090–2095. [Google Scholar] [CrossRef] [Green Version]
- Tesfaye, A.; Di Cello, F.; Hillion, J.; Ronnett, B.M.; Elbahloul, O.; Ashfaq, R.; Dhara, S.; Prochownik, E.; Tworkoski, K.; Reeves, R.; et al. The High-Mobility Group A1 Gene Up-Regulates Cyclooxygenase 2 Expression in Uterine Tumorigenesis. Cancer Res. 2007, 67, 3998–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillion, J.; Smail, S.; Di Cello, F. The HMGA1-COX-2 axis: A key molecular pathway and potential target in pancreatic adenocarcinoma. Pancreatology 2012, 12, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.S.; Xu, Q.; Schmedtje, J.F. Hypoxia induces high-mobility-group protein I(Y) and transcription of the cyclooxygenase-2 gene in human vascular endothelium. Circ. Res. 1998, 83, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, R.M.; Lopez-Guzman, S.; Riascos, D.F.; Macias, A.A.; Layne, M.D.; Cheng, G.; Harris, C.; Chung, S.W.; Reeves, R.; von Andrian, U.H.; et al. Distamycin A inhibits HMGA1-binding to the P-selectin promoter and attenuates lung and liver inflammation during murine endotoxemia. PLoS ONE 2010, 5, e10656. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342. [Google Scholar] [CrossRef] [Green Version]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudenaarden, V.; et al. Plasticity between Epithelial and Mesenchymal Report Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Belton, A.; Gabrovsky, A.; Bae, Y.K.; Reeves, R.; Iacobuzio-Donahue, C.; Huso, D.L.; Resar, L.M.S. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS ONE 2012, 7, e30034. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, S.; Ros, G.; Piazza, S.R.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.H.; Shi, Y.; Tian, Y.; Cao, T.T.; Mao, Y.; Tang, T.Y. HMGA1 accelerates the malignant progression of gastric cancer through stimulating EMT. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3642–3647. [Google Scholar] [CrossRef]
- Shah, S.N.; Cope, L.; Poh, W.; Belton, A.; Roy, S. HMGA1: A Master Regulator of Tumor Progression in Triple-Negative Breast Cancer Cells. PLoS ONE 2013, 8, e63419. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Eaton, E.; Li, S.; Chaffer, C.; Reinhardt, F.; Kah, K.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Guen, V.J.; Chavarria, T.E.; Kröger, C.; Ye, X.; Weinberg, R.A.; Lees, J.A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E10532–E10539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, S.; Hutchins, E.J.; Maruszko, K.; Park, J.H.; Thomson, M.; Bronner, M.E. Bimodal function of chromatin remodeler Hmga1 in neural crest induction and Wnt- dependent emigration. Elife 2020, 9, e57779. [Google Scholar] [CrossRef] [PubMed]
- Bronner, M.E.; LeDouarin, N.M. Development and evolution of the neural crest: An overview. Dev. Biol. 2012, 366, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, S.; Johnston, S.; Johnston, S. PARtitioning Numb. EMBO Rep. 2007, 8, 233–235. [Google Scholar] [CrossRef] [Green Version]
- Puca, F.; Tosti, N.; Federico, A.; Kuzay, Y.; Pepe, A.; Morlando, S.; Savarese, T.; D’Alessio, F.; Colamaio, M.; Sarnataro, D.; et al. HMGA1 negatively regulates NUMB expression at transcriptional and post transcriptional level in glioblastoma stem cells. Cell Cycle 2019, 18, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Donato, R. HMGB1-RAGE regulates muscle satellite cell homeostasis through p38-MAPK- and myogenindependent repression of Pax7 transcription. J. Cell Sci. 2012, 125, 1440–1454. [Google Scholar] [CrossRef] [Green Version]
- Rashmi, R.; Nitish, J.; Satyendra Kumar, S.; Sunita, S.; Vivek, R. Lysophosphatidic acid-RAGE axis promotes lung and mammary oncogenesis via protein kinase B and regulating tumor microenvironment. Cell Commun. Signal. 2020, 18, 170. [Google Scholar] [CrossRef]
- Yin, C.; Li, H.; Zhang, B.; Liu, Y.; Lu, G.; Lu, S.; Sun, L.; Qi, Y.; Li, X.; Chen, W. RAGE-binding S100A8/A9 promotes the migration and invasion of human breast cancer cells through actin polymerization and epithelial-mesenchymal transition. Breast Cancer Res. Treat. 2013, 142, 297–309. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, G.; Sun, R.; Pan, X.; Wang, X.; Li, H.; Sun, Y. miR-185-5p inhibits F-actin polymerization and reverses epithelial mesenchymal transition of human breast cancer cells by modulating RAGE. Mol. Med. Rep. 2018, 2621–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shao, S.; Han, D.; Xu, Y.; Jiao, D.; Wu, J.; Yang, F.; Ge, Y.; Shi, S.; Li, Y.; et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-KB signaling pathway. Int. J. Oncol. 2018, 53, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Könnecke, M.; Burmeister, M.; Pries, R.; Böscke, R.; Bruchhage, K.; Ungefroren, H.; Klimek, L.; Wollenberg, B. Epithelial-Mesenchymal Transition in Chronic Rhinosinusitis: Differences Revealed Between Epithelial Cells from Nasal Polyps and Inferior Turbinates. Arch. Immunol. Ther. Exp. (Warsz). 2017, 65, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Statt, S.; Wu, R.; Chang, H.-T.; Liao, J.-W.; Wang, C.-N.; Shyu, W.-C.; Lee, C.-C. High mobility group box 1-induced epithelial mesenchymal transition in human airway epithelial cells. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.; Liu, H.; Zhang, D.; Liu, Y.; Wang, C.; Liu, F.; Chen, J. HMGB1 Enhances the AGE-Induced Expression of CTGF and TGF-β via RAGE-Dependent Signaling in Renal Tubular Epithelial Cells. Am. J. Nephrol. 2015, 41, 257–266. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pujals, M.; Resar, L.; Villanueva, J. HMGA1, Moonlighting Protein Function, and Cellular Real Estate: Location, Location, Location! Biomolecules 2021, 11, 1334. https://doi.org/10.3390/biom11091334
Pujals M, Resar L, Villanueva J. HMGA1, Moonlighting Protein Function, and Cellular Real Estate: Location, Location, Location! Biomolecules. 2021; 11(9):1334. https://doi.org/10.3390/biom11091334
Chicago/Turabian StylePujals, Mireia, Linda Resar, and Josep Villanueva. 2021. "HMGA1, Moonlighting Protein Function, and Cellular Real Estate: Location, Location, Location!" Biomolecules 11, no. 9: 1334. https://doi.org/10.3390/biom11091334
APA StylePujals, M., Resar, L., & Villanueva, J. (2021). HMGA1, Moonlighting Protein Function, and Cellular Real Estate: Location, Location, Location! Biomolecules, 11(9), 1334. https://doi.org/10.3390/biom11091334