
Accurate Sequence-Based Prediction of Deleterious nsSNPs with Multiple Sequence Profiles and Putative Binding Residues


 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
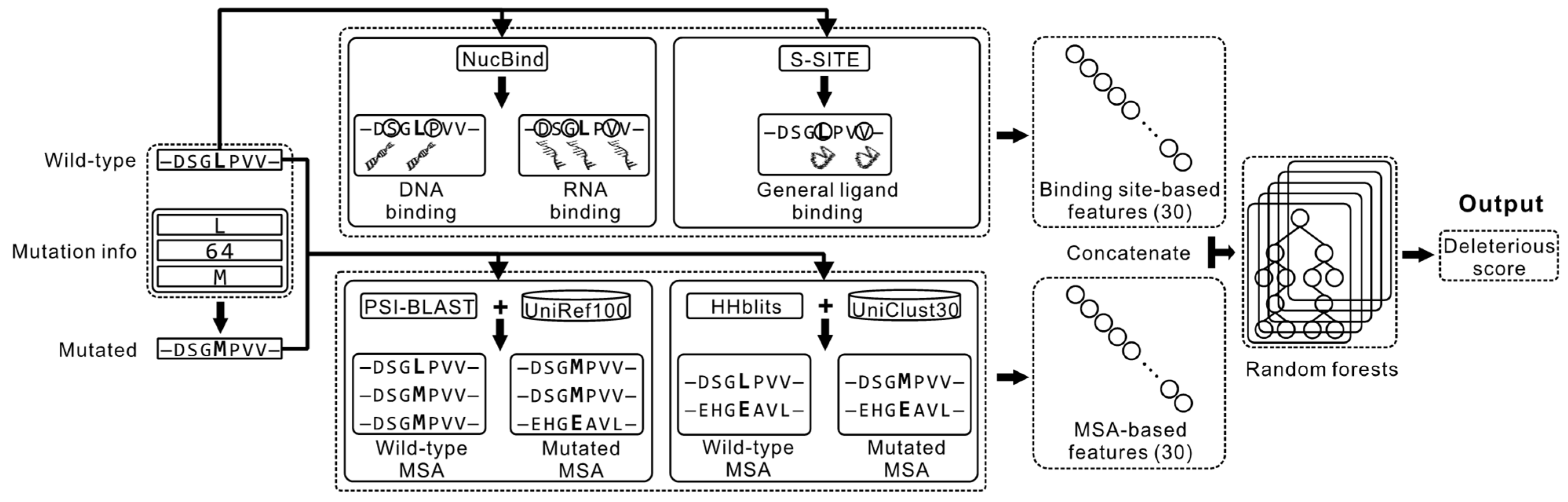
2.1. Overview of the Proposed Method
2.2. Feature Extraction
2.2.1. Features Computed from the MSA Profiles
2.2.2. Features Computed from the Sequence-Based Prediction of the Ligand-Binding Residues
2.3. Benchmark Datasets
2.4. Performance Evaluation
3. Results and Discussion
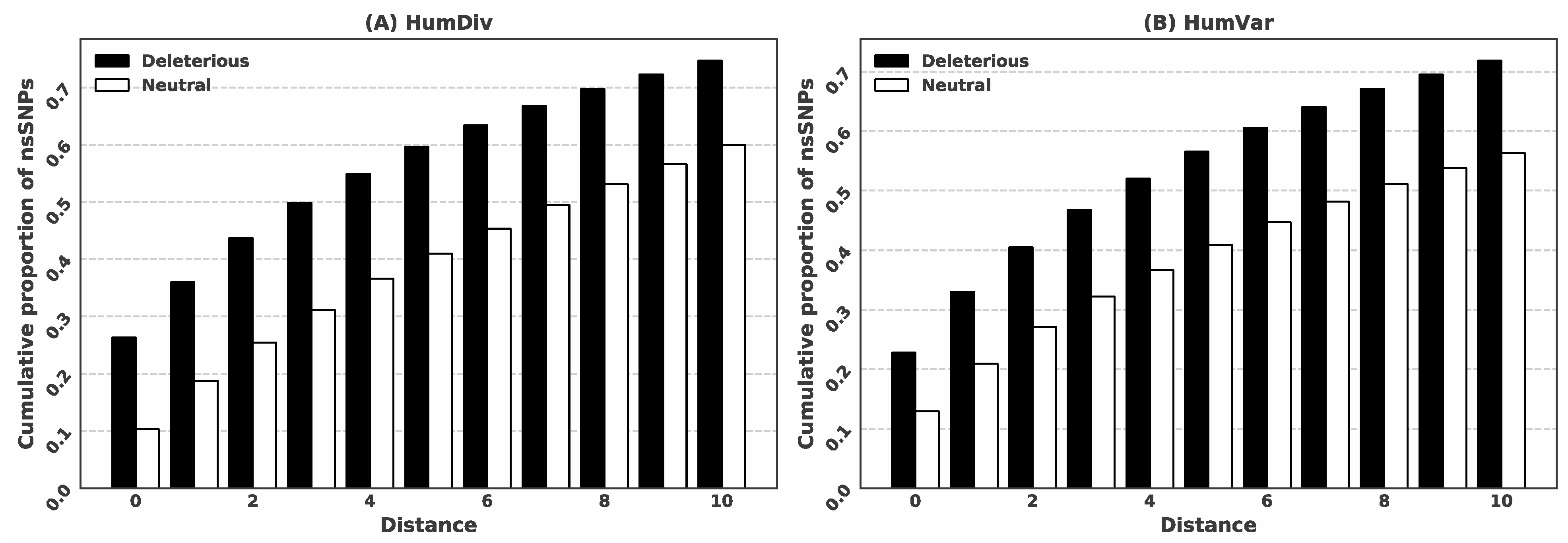
3.1. Deleterious nsSNPs Coincide with the Location of the Putative Ligand-Binding Residues
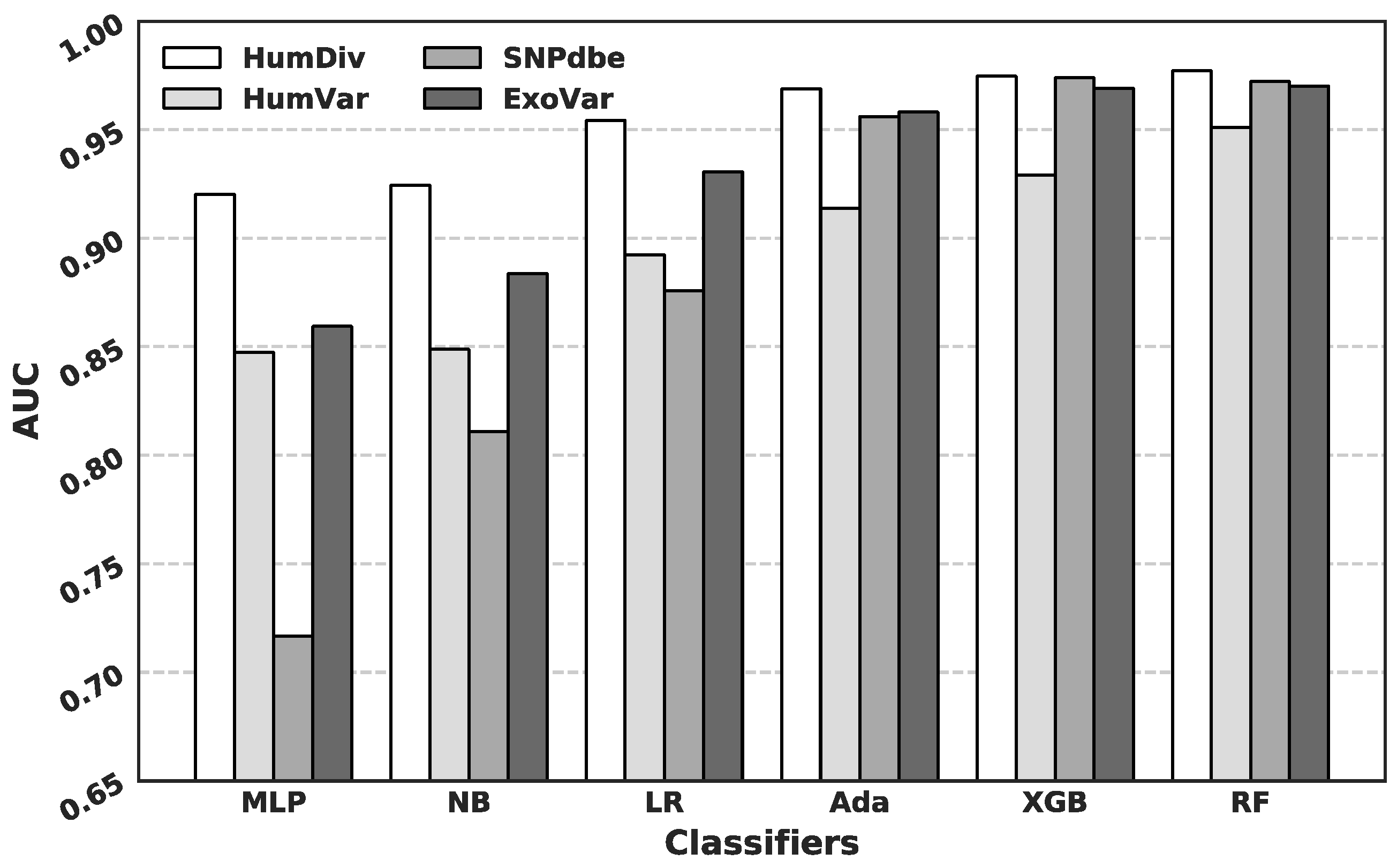
3.2. Comparison of Predictive Performance Generated Using Different Machine Learning Algorithms
3.3. Comparison with Current Predictors on the HumDiv and the HumVar Datasets
3.4. Comparison with Current Predictors on the SNPdbe and ExoVar Datasets
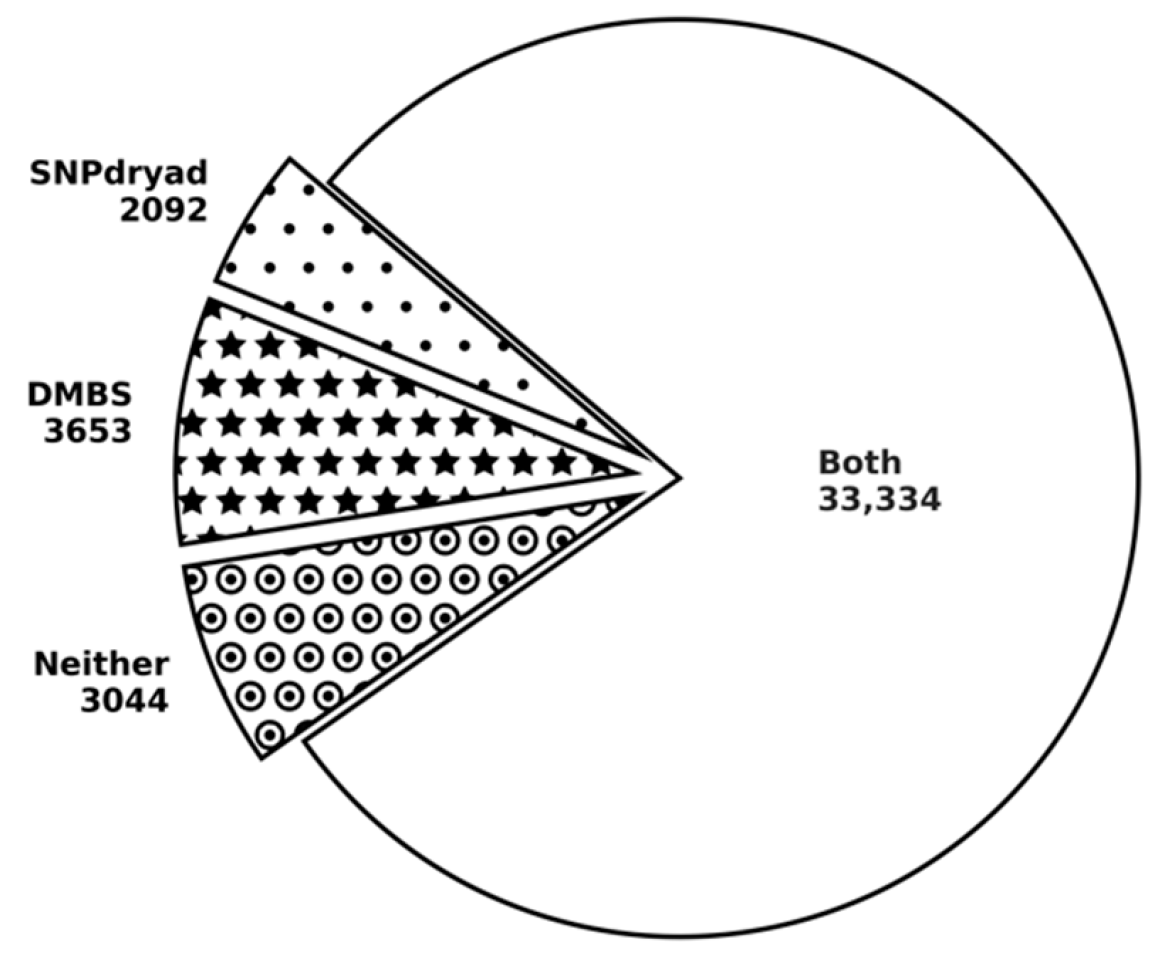
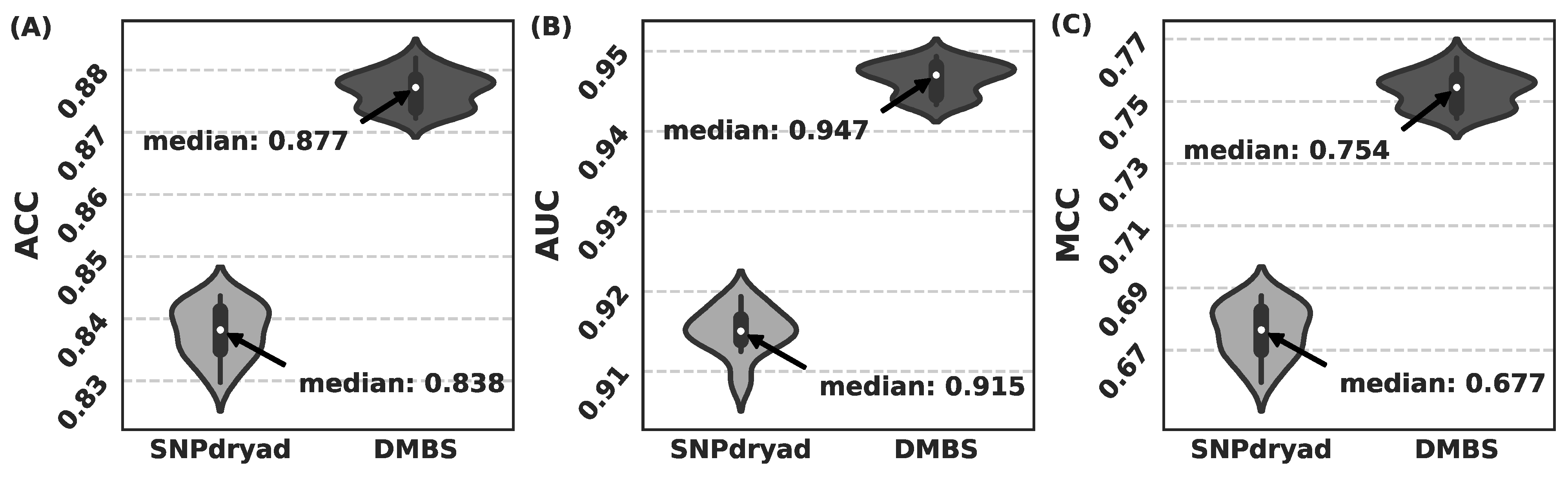
3.5. Comparison with SNPdryad Based on Independent Test
3.6. DMBS Web Server
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wohlrab, H. The human mitochondrial transport/carrier protein family. Nonsynonymous single nucleotide polymorphisms (nsSNPs) and mutations that lead to human diseases. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1263–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Castejon, M.; Marin, F.; Soler-Rivas, C.; Reglero, G.; Visioli, F.; Rodriguez-Casado, A. Functional non-synonymous polymorphisms prediction methods: Current approaches and future developments. Curr. Med. Chem. 2011, 18, 5095–5103. [Google Scholar] [CrossRef] [Green Version]
- Yates, C.M.; Sternberg, M. The Effects of Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) on Protein–Protein Interactions. J. Mol. Biol. 2013, 425, 3949–3963. [Google Scholar] [CrossRef]
- Gfeller, D.; Ernst, A.; Jarvik, N.; Sidhu, S.S.; Bader, G.D. Prediction and Experimental Characterization of nsSNPs Altering Human PDZ-Binding Motifs. PLoS ONE 2014, 9, e94507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conseil, G.; Cole, S. Two Polymorphic Variants of ABCC1 Selectively Alter Drug Resistance and Inhibitor Sensitivity of the Multidrug and Organic Anion Transporter Multidrug Resistance Protein 1. Drug Metab. Dispos. 2013, 41, 2187–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, L.; Fukushima, H.; Karchin, R.; Gow, J.M.; Chinn, L.; Pieper, U.; Segal, M.R.; Kroetz, D.L.; Sali, A. Functional hot spots in human ATP-binding cassette transporter nucleotide binding domains. Protein Sci. 2010, 19, 2110–2121. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, C.; Meier, A.; Rost, B.; Bromberg, Y. SNPdbe: Constructing an nsSNP functional impacts database. Bioinformatics 2011, 28, 601–602. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A Database of Human Non-synonymous SNVs and Their Functional Predictions and Annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.D.; Handsaker, R.E.; Pulit, S.L.; Nizzari, M.M.; O’Donnell, C.J.; de Bakker, P.I.W. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 2008, 24, 2938–2939. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Chem. Biol. 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.-X.; Kwan, J.S.H.; Bao, S.-Y.; Yang, W.; Ho, S.-L.; Song, Y.-Q.; Sham, P.C. Predicting Mendelian Disease-Causing Non-Synonymous Single Nucleotide Variants in Exome Sequencing Studies. PLoS Genet. 2013, 9, e1003143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-C.; Zhang, Z. SNPdryad: Predicting deleterious non-synonymous human SNPs using only orthologous protein sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Hopf, T.A.; Ingraham, J.B.; Poelwijk, F.J.; Schärfe, C.P.; Springer, M.; Sander, C.; Marks, D.S. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 2017, 35, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Liu, M.; Sun, S.; Peng, Z.; Yang, J. Improving the prediction of protein–nucleic acids binding residues via multiple sequence profiles and the consensus of complementary methods. Bioinformatics 2018, 35, 930–936. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, Z.; Yang, J. Improving Sequence-Based Prediction of Protein–Peptide Binding Residues by Introducing Intrinsic Disorder and a Consensus Method. J. Chem. Inf. Model. 2018, 58, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Kurgan, L. Sequence Similarity Searching. Curr. Protoc. Protein Sci. 2018, 95, e71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Deleterious nsSNPs | Neutral nsSNPs | Ratio of Positives | Reference |
|---|---|---|---|---|
| HumVar | 20,985 | 21,138 | 49.82% | [13] |
| HumDiv | 5322 | 7070 | 42.95% | [13] |
| SNPdbe | 2263 | 307 | 88.05% | [9] |
| ExoVar | 4567 | 3145 | 59.22% | [15] |
| Method | HumDiv | HumVar |
|---|---|---|
| SIFT | 0.91 | 0.86 |
| PolyPhen2 | 0.95 | 0.89 |
| SNPdryad | 0.98 | 0.91 |
| DMBS_BR | 0.908 | 0.909 |
| DMBS_MSA | 0.976 | 0.941 |
| DMBS | 0.977 | 0.948 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, R.; Cao, B.; Peng, Z.; Oldfield, C.J.; Kurgan, L.; Wong, K.-C.; Yang, J. Accurate Sequence-Based Prediction of Deleterious nsSNPs with Multiple Sequence Profiles and Putative Binding Residues. Biomolecules 2021, 11, 1337. https://doi.org/10.3390/biom11091337
Song R, Cao B, Peng Z, Oldfield CJ, Kurgan L, Wong K-C, Yang J. Accurate Sequence-Based Prediction of Deleterious nsSNPs with Multiple Sequence Profiles and Putative Binding Residues. Biomolecules. 2021; 11(9):1337. https://doi.org/10.3390/biom11091337
Chicago/Turabian StyleSong, Ruiyang, Baixin Cao, Zhenling Peng, Christopher J. Oldfield, Lukasz Kurgan, Ka-Chun Wong, and Jianyi Yang. 2021. "Accurate Sequence-Based Prediction of Deleterious nsSNPs with Multiple Sequence Profiles and Putative Binding Residues" Biomolecules 11, no. 9: 1337. https://doi.org/10.3390/biom11091337
APA StyleSong, R., Cao, B., Peng, Z., Oldfield, C. J., Kurgan, L., Wong, K. -C., & Yang, J. (2021). Accurate Sequence-Based Prediction of Deleterious nsSNPs with Multiple Sequence Profiles and Putative Binding Residues. Biomolecules, 11(9), 1337. https://doi.org/10.3390/biom11091337