
TGFβ-Neurotrophin Interactions in Heart, Retina, and Brain


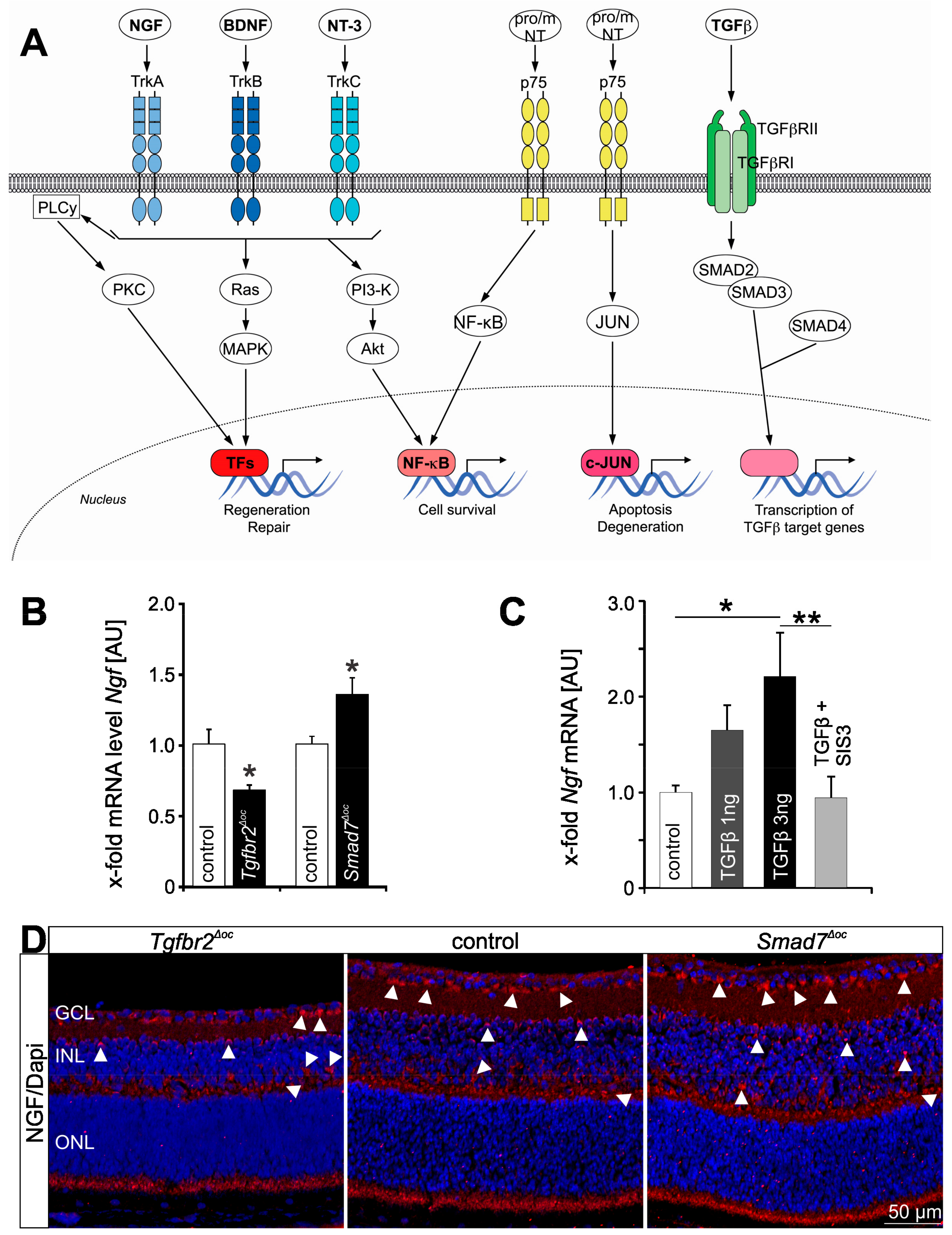{kind=link}
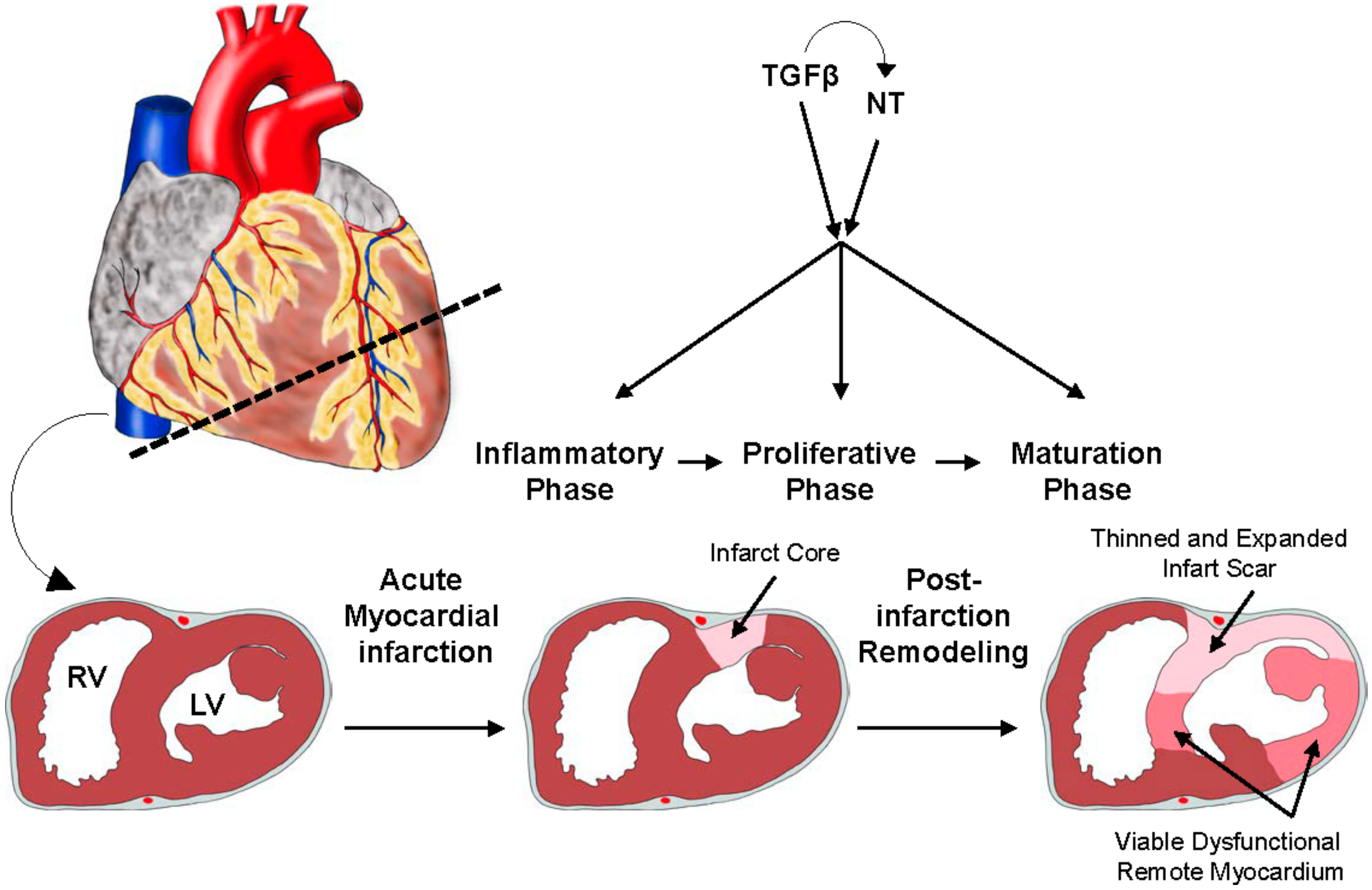{kind=link}
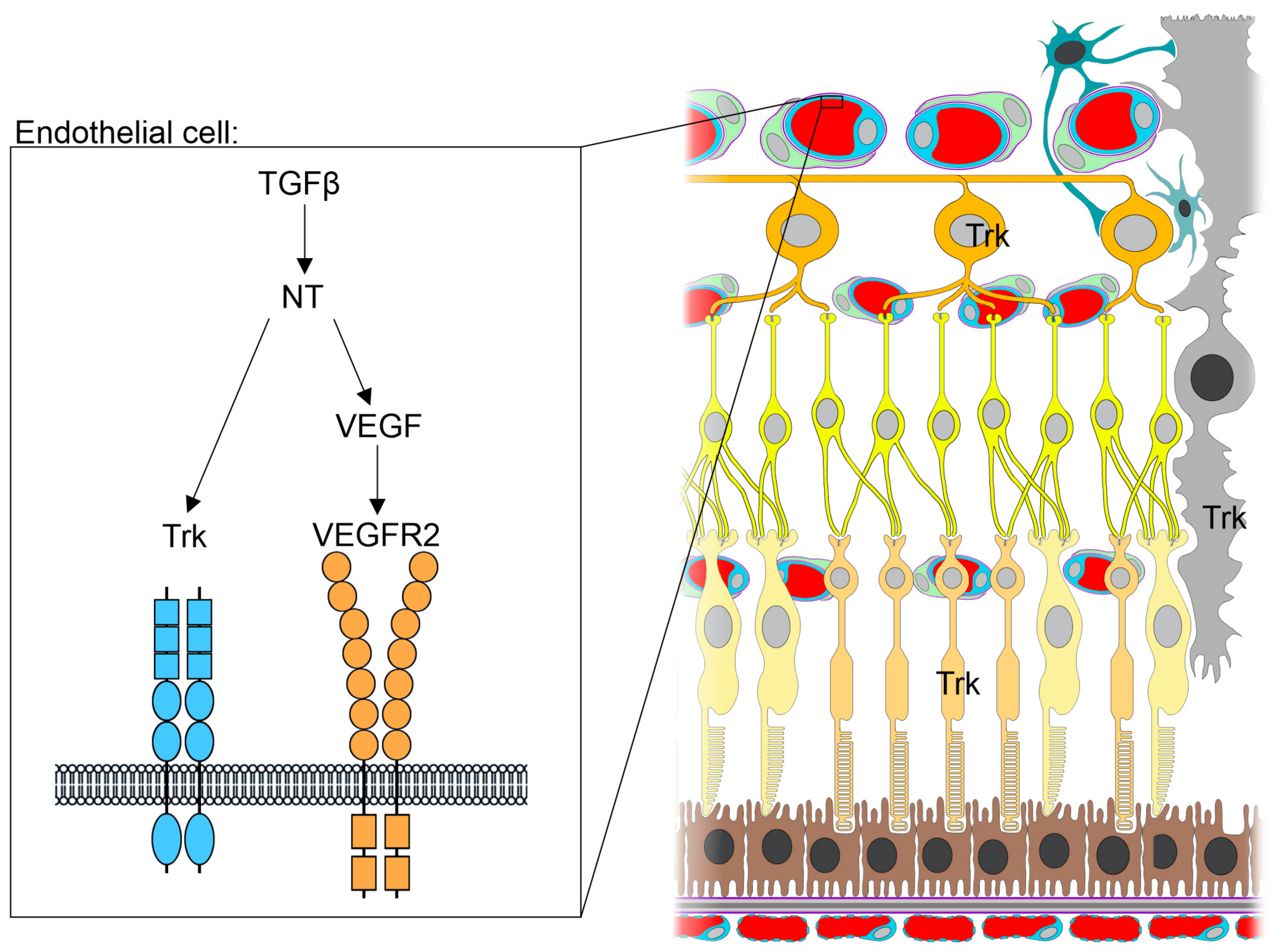{kind=link}
{kind=link}
{kind=link}
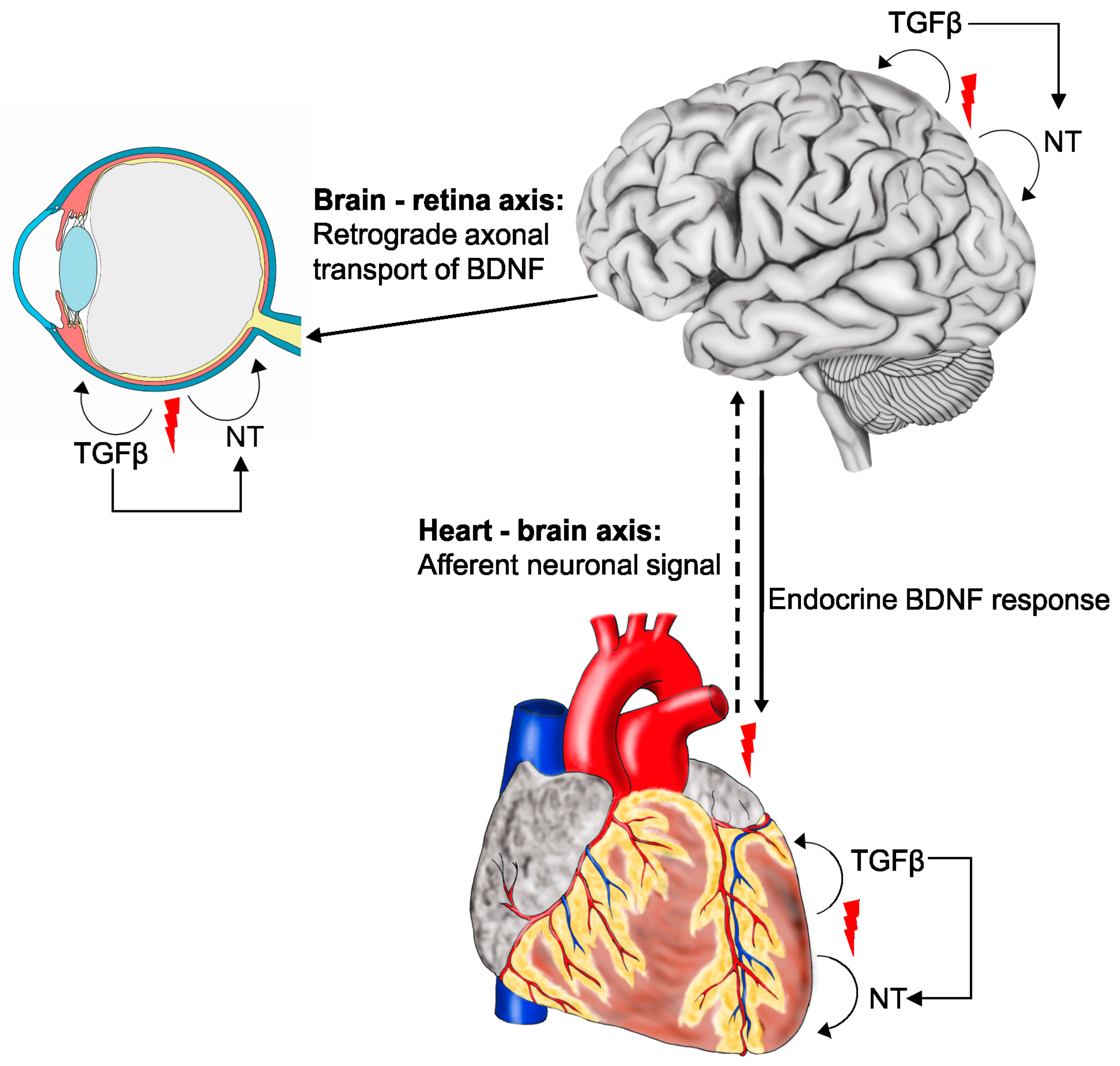{kind=link}
Abstract
:1. Introduction

2. TGFβ and Neurotrophin Signaling in the Heart
2.1. Neurotrophins in the Heart
2.2. TGFβ Signaling in the Cardiovascular System
2.3. A Brief Overview: Ischemic Heart Disease and Myocardial Infarction (MI)
2.4. Neurotrophins in MI
2.5. Cell Type-Specific Effects of Neurotrophins and the Heart-Brain Axis
2.6. The Role of TGFβ Signaling in MI: Timing Matters
2.7. Specific Actions of TGFβ Signaling in MI: Cell-Type Matters
3. TGFβ and Neurotrophin Signaling in the Central Nervous System (CNS): Retina
3.1. Neurotrophins in the Retina and the Brain-Retina Axis
3.2. TGF-β Signaling and Its Cross-Talk with Neurotrophins in the Eye
3.3. A Brief Overview: (Neo)Vascular Eeye Diseases
3.4. Neurotrophins in (Neo)Vascular Eye Diseases
3.5. TGF-β Signaling in (Neo)Vascular Eye Diseases
4. TGFβ and Neurotrophin Signaling in the Central Nervous System (CNS): Brain
4.1. Neurotrophins in Nervous System Development and Homeostasis
4.2. TGFβ Signaling in the Brain
4.3. A Brief Overview: Ischemic Stroke
4.4. Neurotrophins and Ischemic Stroke
4.5. TGFβ and Ischemic Stroke
4.6. TGFβ-Neurotrophin Cross-Talk in the Brain
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DR | diabetic retinopathy |
| AMD | age-related macular degeneration |
| TGFβ | transforming growth factor β |
| CNS/PNS | central and peripheral nervous systems, |
| BDNF | Brain-derived neurotrophic factor |
| NGF | nerve growth factor |
| NT-3 | neurotrophin-3 |
| NT-4/5 | neurotrophin-4/5 |
| TrkA–C | tropomyosin receptor kinase A–C |
| p75NTR | neurotrophin receptor p75 |
| TGFβR1/2 | TGFβ receptor types 1 and 2 |
| BMP | bone morphogenetic protein |
| GDF | growth differentiation factors |
| ALK1/5 | activin A receptor like type 1/5 |
| SMAD | abbreviation refers to “small worm phenotype” (SMA) and “mother against decapentaplegic” (MAD), which are homologs of C. elegans and drosophila |
| ECM | extracellular matrix |
| MI | myocardial infraction |
| BAX | BCL2 Associated X, Apoptosis Regulator |
| BCL2 | BCL2 Apoptosis Regulator |
| VEGF | vascular endothelial growth factor |
| CAP | capsaicin |
| GW788388 | small-molecule inhibitor of TGFβR1 |
| BRB | blood-retinal barrier |
| BBB | blood-brain barrier |
| RPE | retinal pigment epithelium |
| RGC | retinal ganglion cells |
| CNV | choroidal neovascularization |
| tPA | tissue plasminogen activator |
| NSC | neuronal stem cells |
| t/pMCAO | transient or permanent middle cerebral artery occlusion |
| eNOS | endothelial nitric oxide synthase |
| ENG | endoglin |
References
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Al Suwaidi, S.; AlKatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.; Alzaabi, M.E.H.; Al Darmaki, R.S.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef]
- Blindness, G.B.D.; Vision Impairment Collaborators; Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Klein, B.E. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol. 2007, 14, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Yokoyama, M.; Toko, H.; Tateno, K.; Moriya, J.; Shimizu, I.; Nojima, A.; Ito, T.; Yoshida, Y.; Kobayashi, Y.; et al. Brain-derived neurotrophic factor protects against cardiac dysfunction after myocardial infarction via a central nervous system-mediated pathway. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1902–1909. [Google Scholar] [CrossRef] [Green Version]
- Claes, M.; De Groef, L.; Moons, L. Target-Derived Neurotrophic Factor Deprivation Puts Retinal Ganglion Cells on Death Row: Cold Hard Evidence and Caveats. Int. J. Mol. Sci. 2019, 20, 4314. [Google Scholar] [CrossRef] [Green Version]
- Ola, M.S.; Nawaz, M.I.; El-Asrar, A.A.; Abouammoh, M.; Alhomida, A.S. Reduced levels of brain derived neurotrophic factor (BDNF) in the serum of diabetic retinopathy patients and in the retina of diabetic rats. Cell. Mol. Neurobiol. 2013, 33, 359–367. [Google Scholar] [CrossRef]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Vilahur, G.; Juan-Babot, O.; Pena, E.; Onate, B.; Casani, L.; Badimon, L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 522–533. [Google Scholar] [CrossRef]
- Telegina, D.V.; Kolosova, N.G.; Kozhevnikova, O.S. Immunohistochemical localization of NGF, BDNF, and their receptors in a normal and AMD-like rat retina. BMC Med. Genom. 2019, 12, 48. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Mohammad, G.; De Hertogh, G.; Nawaz, M.I.; Van Den Eynde, K.; Siddiquei, M.M.; Struyf, S.; Opdenakker, G.; Geboes, K. Neurotrophins and neurotrophin receptors in proliferative diabetic retinopathy. PLoS ONE 2013, 8, e65472. [Google Scholar] [CrossRef]
- Kokaia, Z.; Andsberg, G.; Yan, Q.; Lindvall, O. Rapid alterations of BDNF protein levels in the rat brain after focal ischemia: Evidence for increased synthesis and anterograde axonal transport. Exp. Neurol. 1998, 154, 289–301. [Google Scholar] [CrossRef]
- Kokaia, Z.; Zhao, Q.; Kokaia, M.; Elmer, E.; Metsis, M.; Smith, M.L.; Siesjo, B.K.; Lindvall, O. Regulation of brain-derived neurotrophic factor gene expression after transient middle cerebral artery occlusion with and without brain damage. Exp. Neurol. 1995, 136, 73–88. [Google Scholar] [CrossRef]
- Pál, G.; Lovas, G.; Dobolyi, A. Induction of Transforming Growth Factor Beta Receptors following Focal Ischemia in the Rat Brain. PLoS ONE 2014, 9, e106544. [Google Scholar] [CrossRef] [Green Version]
- Krupinski, J.; Kumar, P.; Kumar, S.; Kaluza, J. Increased Expression of TGF-β1 in Brain Tissue After Ischemic Stroke in Humans. Stroke 1996, 27, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. 2012, 846, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.M.; Barde, Y.A. Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature 1988, 331, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Levi-Montalcini, R.; Hamburger, V. A Nerve Growth-Stimulating Factor Isolated from Sarcom as 37 and 180. Proc. Natl. Acad. Sci. USA 1954, 40, 1014–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi-Montalcini, R.; Hamburger, V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 1951, 116, 321–361. [Google Scholar] [CrossRef] [PubMed]
- Pius-Sadowska, E.; Machalinski, B. BDNF—A key player in cardiovascular system. J. Mol. Cell. Cardiol. 2017, 110, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Bahls, M.; Konemann, S.; Markus, M.R.P.; Wenzel, K.; Friedrich, N.; Nauck, M.; Volzke, H.; Steveling, A.; Janowitz, D.; Grabe, H.J.; et al. Brain-derived neurotrophic factor is related with adverse cardiac remodeling and high NTproBNP. Sci. Rep. 2019, 9, 15421. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.R.; Reichardt, L.F. Molecular cloning of a human gene that is a member of the nerve growth factor family. Proc. Natl. Acad. Sci. USA 1990, 87, 8060–8064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ip, N.Y.; Ibanez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Le Beau, M.M.; Espinosa, R., 3rd; Squinto, S.P.; et al. Mammalian neurotrophin-4: Structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef] [Green Version]
- Caporali, A.; Emanueli, C. Cardiovascular actions of neurotrophins. Physiol. Rev. 2009, 89, 279–308. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Lanahan, A.; Buck, C.R.; Sehgal, A.; Morgan, C.; Mercer, E.; Bothwell, M.; Chao, M. Expression and structure of the human NGF receptor. Cell 1986, 47, 545–554. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Hempstead, B.L.; Martin-Zanca, D.; Chao, M.V.; Parada, L.F. The trk proto-oncogene product: A signal transducing receptor for nerve growth factor. Science 1991, 252, 554–558. [Google Scholar] [CrossRef]
- Klein, R.; Jing, S.Q.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Gibon, J.; Barker, P.A. Neurotrophins and Proneurotrophins: Focus on Synaptic Activity and Plasticity in the Brain. Neuroscientist 2017, 23, 587–604. [Google Scholar] [CrossRef]
- Garcia, T.B.; Hollborn, M.; Bringmann, A. Expression and signaling of NGF in the healthy and injured retina. Cytokine Growth Factor Rev. 2017, 34, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Krieglstein, K.; Strelau, J.; Schober, A.; Sullivan, A.; Unsicker, K. TGF-beta and the regulation of neuron survival and death. J. Physiol. Paris 2002, 96, 25–30. [Google Scholar] [CrossRef]
- Kugler, M.; Schlecht, A.; Fuchshofer, R.; Schmitt, S.I.; Kleiter, I.; Aigner, L.; Tamm, E.R.; Braunger, B.M. SMAD7 deficiency stimulates Müller progenitor cell proliferation during the development of the mammalian retina. Histochem. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Schlecht, A.; Leimbeck, S.V.; Jägle, H.; Feuchtinger, A.; Tamm, E.R.; Braunger, B.M. Deletion of Endothelial Transforming Growth Factor-β Signaling Leads to Choroidal Neovascularization. Am. J. Pathol. 2017, 187, 2570–2589. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef]
- Leveen, P.; Larsson, J.; Ehinger, M.; Cilio, C.M.; Sundler, M.; Sjostrand, L.J.; Holmdahl, R.; Karlsson, S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood 2002, 100, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Braunger, B.M.; Leimbeck, S.V.; Schlecht, A.; Volz, C.; Jagle, H.; Tamm, E.R. Deletion of ocular transforming growth factor beta signaling mimics essential characteristics of diabetic retinopathy. Am. J. Pathol. 2015, 185, 1749–1768. [Google Scholar] [CrossRef]
- Braunger, B.M.; Pielmeier, S.; Demmer, C.; Landstorfer, V.; Kawall, D.; Abramov, N.; Leibinger, M.; Kleiter, I.; Fischer, D.; Jagle, H.; et al. TGF-beta signaling protects retinal neurons from programmed cell death during the development of the mammalian eye. J. Neurosci. 2013, 33, 14246–14258. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-beta Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.W.D.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef]
- Roberts, A.B.; Kim, S.J.; Noma, T.; Glick, A.B.; Lafyatis, R.; Lechleider, R.; Jakowlew, S.B.; Geiser, A.; O’Reilly, M.A.; Danielpour, D.; et al. Multiple forms of TGF-beta: Distinct promoters and differential expression. Ciba Found. Symp. 1991, 157, 7–15; discussion 15–28. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-β family in wound healing, burns and scarring: A review. Int. J. Burn. Trauma 2012, 2, 18–28. [Google Scholar]
- Flaumenhaft, R.; Abe, M.; Sato, Y.; Miyazono, K.; Harpel, J.; Heldin, C.H.; Rifkin, D.B. Role of the latent TGF-beta binding protein in the activation of latent TGF-beta by co-cultures of endothelial and smooth muscle cells. J. Cell Biol. 1993, 120, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sakai, T. Biological Significance of Local TGF-beta Activation in Liver Diseases. Front. Physiol. 2012, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Sometani, A.; Kataoka, H.; Nitta, A.; Fukumitsu, H.; Nomoto, H.; Furukawa, S. Transforming growth factor-beta1 enhances expression of brain-derived neurotrophic factor and its receptor, TrkB, in neurons cultured from rat cerebral cortex. J. Neurosci. Res. 2001, 66, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Silverman, S.M.; Zhao, L.; Villasmil, R.; Campos, M.M.; Amaral, J.; Wong, W.T. Absence of TGFbeta signaling in retinal microglia induces retinal degeneration and exacerbates choroidal neovascularization. eLife 2019, 8, e42049. [Google Scholar] [CrossRef]
- Krieglstein, K.; Unsicker, K. Distinct modulatory actions of TGF-beta and LIF on neurotrophin-mediated survival of developing sensory neurons. Neurochem. Res. 1996, 21, 843–850. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Klein, R.; Schnapp, A.; Long, L.K.; Bryant, S.; Lewin, A.; Lira, S.A.; Barbacid, M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 1994, 368, 246–249. [Google Scholar] [CrossRef]
- Crowley, C.; Spencer, S.D.; Nishimura, M.C.; Chen, K.S.; Pitts-Meek, S.; Armanini, M.P.; Ling, L.H.; McMahon, S.B.; Shelton, D.L.; Levinson, A.D.; et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 1994, 76, 1001–1011. [Google Scholar] [CrossRef]
- Sendtner, M.; Holtmann, B.; Kolbeck, R.; Thoenen, H.; Barde, Y.A. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature 1992, 360, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Silos-Santiago, I.; Smeyne, R.J.; Lira, S.A.; Brambilla, R.; Bryant, S.; Zhang, L.; Snider, W.D.; Barbacid, M. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature 1994, 368, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Smeyne, R.J.; Wurst, W.; Long, L.K.; Auerbach, B.A.; Joyner, A.L.; Barbacid, M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 1993, 75, 113–122. [Google Scholar] [CrossRef]
- Ernfors, P.; Lee, K.F.; Kucera, J.; Jaenisch, R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell 1994, 77, 503–512. [Google Scholar] [CrossRef]
- Jones, K.R.; Farinas, I.; Backus, C.; Reichardt, L.F. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 1994, 76, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Ieda, M.; Kanazawa, H.; Ieda, Y.; Kimura, K.; Matsumura, K.; Tomita, Y.; Yagi, T.; Onizuka, T.; Shimoji, K.; Ogawa, S.; et al. Nerve growth factor is critical for cardiac sensory innervation and rescues neuropathy in diabetic hearts. Circulation 2006, 114, 2351–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, M.J.; Miranda, R.C.; Kraemer, R.; McCaffrey, T.A.; Tessarollo, L.; Mahadeo, D.; Sharif, S.; Kaplan, D.R.; Tsoulfas, P.; Parada, L.; et al. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells. Regulation of expression in response to injury. Am. J. Pathol. 1995, 147, 309–324. [Google Scholar]
- Scarisbrick, I.A.; Jones, E.G.; Isackson, P.J. Coexpression of mRNAs for NGF, BDNF, and NT-3 in the cardiovascular system of the pre- and postnatal rat. J. Neurosci. 1993, 13, 875–893. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.O.; Laurikainen, A.; Vakeva, A.; Meri, S.; Saarma, M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J. Pathol. 2001, 194, 247–253. [Google Scholar] [CrossRef]
- Hiltunen, J.O.; Arumae, U.; Moshnyakov, M.; Saarma, M. Expression of mRNAs for neurotrophins and their receptors in developing rat heart. Circ. Res. 1996, 79, 930–939. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D.; Theres, H.; Englert, C.; Schedl, A.; Scholz, H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 2005, 19, 2631–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, M.J.; Lin, M.I.; Wiegn, P.; Ringstedt, T.; Kraemer, R.; Hahn, R.; Wang, S.; Ibanez, C.F.; Rafii, S.; Hempstead, B.L. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development 2000, 127, 4531–4540. [Google Scholar] [CrossRef]
- Hassankhani, A.; Steinhelper, M.E.; Soonpaa, M.H.; Katz, E.B.; Taylor, D.A.; Andrade-Rozental, A.; Factor, S.M.; Steinberg, J.J.; Field, L.J.; Federoff, H.J. Overexpression of NGF within the heart of transgenic mice causes hyperinnervation, cardiac enlargement, and hyperplasia of ectopic cells. Dev. Biol. 1995, 169, 309–321. [Google Scholar] [CrossRef]
- Anastasia, A.; Deinhardt, K.; Wang, S.; Martin, L.; Nichol, D.; Irmady, K.; Trinh, J.; Parada, L.; Rafii, S.; Hempstead, B.L.; et al. Trkb signaling in pericytes is required for cardiac microvessel stabilization. PLoS ONE 2014, 9, e87406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Euler, G. Good and bad sides of TGFβ-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Thompson, N.L.; Flanders, K.C.; Smith, J.M.; Ellingsworth, L.R.; Roberts, A.B.; Sporn, M.B. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J. Cell Biol. 1989, 108, 661–669. [Google Scholar] [CrossRef]
- Larsson, J.; Goumans, M.J.; Sjostrand, L.J.; van Rooijen, M.A.; Ward, D.; Leveen, P.; Xu, X.; ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urness, L.D.; Sorensen, L.K.; Li, D.Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 2000, 26, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective angiogenesis in mice lacking endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef]
- ten Dijke, P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar] [CrossRef]
- Yang, X.; Castilla, L.H.; Xu, X.; Li, C.; Gotay, J.; Weinstein, M.; Liu, P.P.; Deng, C.X. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 1999, 126, 1571–1580. [Google Scholar] [CrossRef]
- Sridurongrit, S.; Larsson, J.; Schwartz, R.; Ruiz-Lozano, P.; Kaartinen, V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev. Biol. 2008, 322, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Umbarkar, P.; Singh, A.P.; Gupte, M.; Verma, V.K.; Galindo, C.L.; Guo, Y.; Zhang, Q.; McNamara, J.W.; Force, T.; Lal, H. Cardiomyocyte SMAD4-Dependent TGF-beta Signaling is Essential to Maintain Adult Heart Homeostasis. JACC Basic Transl. Sci. 2019, 4, 41–53. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef]
- Reimer, K.A.; Lowe, J.E.; Rasmussen, M.M.; Jennings, R.B. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977, 56, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Rivera Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [Green Version]
- Cochain, C.; Channon, K.M.; Silvestre, J.S. Angiogenesis in the infarcted myocardium. Antioxid. Redox Signal. 2013, 18, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Lahn, A.M.; et al. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and after Myocardial Infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Edgley, A.J.; Krum, H.; Kelly, D.J. Targeting fibrosis for the treatment of heart failure: A role for transforming growth factor-beta. Cardiovasc. Ther. 2012, 30, e30–e40. [Google Scholar] [CrossRef]
- Rainer, P.P.; Hao, S.; Vanhoutte, D.; Lee, D.I.; Koitabashi, N.; Molkentin, J.D.; Kass, D.A. Cardiomyocyte-specific transforming growth factor β suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ. Res. 2014, 114, 1246–1257. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Huang, S.; Su, Y.; Wu, Y.J.; Hanna, A.; Brickshawana, A.; Graff, J.; Frangogiannis, N.G. Macrophage Smad3 Protects the Infarcted Heart, Stimulating Phagocytosis and Regulating Inflammation. Circ. Res. 2019, 125, 55–70. [Google Scholar] [CrossRef]
- Hao, J.; Ju, H.; Zhao, S.; Junaid, A.; Scammell-La Fleur, T.; Dixon, I.M. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J. Mol. Cell. Cardiol. 1999, 31, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.J.; Schroeder, M.E.; Aguado, B.A.; Anseth, K.S.; Leinwand, L.A. Matters of the heart: Cellular sex differences. J. Mol. Cell. Cardiol. 2021, 160, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Halade, G.V.; Ma, Y.; Ramirez, T.A.; Zhang, J.; Dai, Q.; Hensler, J.G.; Lopez, E.F.; Ghasemi, O.; Jin, Y.F.; Lindsey, M.L. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1830–H1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.C.; Dang, Y.Y.; Gao, H.Y.; Wang, Z.T.; Gao, M.; Yang, Y.; Zhang, H.T.; Xu, R.X. Local Injection of Lenti-BDNF at the Lesion Site Promotes M2 Macrophage Polarization and Inhibits Inflammatory Response After Spinal Cord Injury in Mice. Cell. Mol. Neurobiol. 2015, 35, 881–890. [Google Scholar] [CrossRef]
- Sandrini, L.; Castiglioni, L.; Amadio, P.; Werba, J.P.; Eligini, S.; Fiorelli, S.; Zara, M.; Castiglioni, S.; Bellosta, S.; Lee, F.S.; et al. Impact of BDNF Val66Met Polymorphism on Myocardial Infarction: Exploring the Macrophage Phenotype. Cells 2020, 9, 1084. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Martin, K.C.; Jackson, J.K.; Beppu, K.; Woo, C.W.; Thiele, C.J. Brain-derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1alpha in neuroblastoma cells. Cancer Res. 2006, 66, 4249–4255. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Liu, Y.; Zhang, Y.; Chen, Z.Y. Association of plasma brain-derived neurotrophic factor and cardiovascular risk factors and prognosis in angina pectoris. Biochem. Biophys. Res. Commun. 2011, 415, 99–103. [Google Scholar] [CrossRef]
- Fukushima, A.; Kinugawa, S.; Homma, T.; Masaki, Y.; Furihata, T.; Yokota, T.; Matsushima, S.; Abe, T.; Suga, T.; Takada, S.; et al. Decreased serum brain-derived neurotrophic factor levels are correlated with exercise intolerance in patients with heart failure. Int. J. Cardiol. 2013, 168, e142–e144. [Google Scholar] [CrossRef]
- Takashio, S.; Sugiyama, S.; Yamamuro, M.; Takahama, H.; Hayashi, T.; Sugano, Y.; Izumiya, Y.; Hokimoto, S.; Minamino, N.; Yasuda, S.; et al. Significance of low plasma levels of brain-derived neurotrophic factor in patients with heart failure. Am. J. Cardiol. 2015, 116, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Manni, L.; Nikolova, V.; Vyagova, D.; Chaldakov, G.N.; Aloe, L. Reduced plasma levels of NGF and BDNF in patients with acute coronary syndromes. Int. J. Cardiol. 2005, 102, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Tsutsui, H.; Shiomi, T.; Matsusaka, H.; Matsushima, S.; Wen, J.; Kubota, T.; Takeshita, A. Inhibition of TGF-β signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc. Res. 2004, 64, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, S.; Hu, K.; Adamek, A.; Wolf, J.; Sallam, A.; Kg Maier, S.; Lonning, S.; Ling, H.; Ertl, G.; Bauersachs, J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res. Cardiol. 2008, 103, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.F.; Frangogiannis, N.G. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007, 116, 2127–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.M.; Zhang, Y.; Connelly, K.A.; Gilbert, R.E.; Kelly, D.J. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1415–H1425. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Takemura, G.; Kosai, K.-i.; Li, Y.; Takahashi, T.; Esaki, M.; Yuge, K.; Miyata, S.; Maruyama, R.; Mikami, A.; et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation 2005, 111, 2430–2437. [Google Scholar] [CrossRef]
- Shimano, M.; Ouchi, N.; Walsh, K. Cardiokines: Recent progress in elucidating the cardiac secretome. Circulation 2012, 126, e327–e332. [Google Scholar] [CrossRef]
- Hulsmans, M.; Nahrendorf, M. Smad3 Cranks Up the Appetite of Infarct Macrophages. Circ. Res. 2019, 125, 71–73. [Google Scholar] [CrossRef]
- Graw, J. Eye development. Curr. Top. Dev. Biol. 2010, 90, 343–386. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21 (Suppl. 6), S3–S9. [Google Scholar] [CrossRef]
- Karl, M.O.; Hayes, S.; Nelson, B.R.; Tan, K.; Buckingham, B.; Reh, T.A. Stimulation of neural regeneration in the mouse retina. Proc. Natl. Acad. Sci. USA 2008, 105, 19508–19513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.F.; Poche, R.A. Awakening the regenerative potential of the mammalian retina. Development 2019, 146, dev182642. [Google Scholar] [CrossRef] [Green Version]
- Pietrucha-Dutczak, M.; Amadio, M.; Govoni, S.; Lewin-Kowalik, J.; Smedowski, A. The Role of Endogenous Neuroprotective Mechanisms in the Prevention of Retinal Ganglion Cells Degeneration. Front. Neurosci. 2018, 12, 834. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.L.; Zeiler, S.R.; Jones, K.R. Patterned expression of BDNF and NT-3 in the retina and anterior segment of the developing mammalian eye. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2996–3005. [Google Scholar]
- Ma, Y.T.; Hsieh, T.; Forbes, M.E.; Johnson, J.E.; Frost, D.O. BDNF injected into the superior colliculus reduces developmental retinal ganglion cell death. J. Neurosci. 1998, 18, 2097–2107. [Google Scholar] [CrossRef]
- Mansour-Robaey, S.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1994, 91, 1632–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, M.; Horie, H.; Iijima, Y.; Dezawa, M.; Sawada, H.; Ishikawa, Y. Brain-derived neurotrophic factor enhances neurite regeneration from retinal ganglion cells in aged human retina in vitro. Exp. Eye Res. 2002, 74, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Sawai, H.; Fukuda, Y. Survival of axotomized retinal ganglion cells in adult mammals. Clin. Neurosci. 1997, 4, 233–239. [Google Scholar]
- Weibel, D.; Kreutzberg, G.W.; Schwab, M.E. Brain-derived neurotrophic factor (BDNF) prevents lesion-induced axonal die-back in young rat optic nerve. Brain Res. 1995, 679, 249–254. [Google Scholar] [CrossRef]
- Hackam, A.S. Regulation of neurotrophin expression and activity in the retina. Adv. Exp. Med. Biol. 2008, 613, 343–349. [Google Scholar] [CrossRef]
- Vecino, E.; Garcia-Crespo, D.; Garcia, M.; Martinez-Millan, L.; Sharma, S.C.; Carrascal, E. Rat retinal ganglion cells co-express brain derived neurotrophic factor (BDNF) and its receptor TrkB. Vis. Res. 2002, 42, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Okoye, G.; Zimmer, J.; Sung, J.; Gehlbach, P.; Deering, T.; Nambu, H.; Hackett, S.; Melia, M.; Esumi, N.; Zack, D.J.; et al. Increased expression of brain-derived neurotrophic factor preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J. Neurosci. 2003, 23, 4164–4172. [Google Scholar] [CrossRef]
- Vecino, E.; Caminos, E.; Ugarte, M.; Martin-Zanca, D.; Osborne, N.N. Immunohistochemical distribution of neurotrophins and their receptors in the rat retina and the effects of ischemia and reperfusion. Gen. Pharm. 1998, 30, 305–314. [Google Scholar] [CrossRef]
- DiStefano, P.S.; Friedman, B.; Radziejewski, C.; Alexander, C.; Boland, P.; Schick, C.M.; Lindsay, R.M.; Wiegand, S.J. The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron 1992, 8, 983–993. [Google Scholar] [CrossRef]
- Martin, K.R.; Quigley, H.A.; Zack, D.J.; Levkovitch-Verbin, H.; Kielczewski, J.; Valenta, D.; Baumrind, L.; Pease, M.E.; Klein, R.L.; Hauswirth, W.W. Gene therapy with brain-derived neurotrophic factor as a protection: Retinal ganglion cells in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4357–4365. [Google Scholar] [CrossRef] [PubMed]
- Vrabec, J.P.; Levin, L.A. The neurobiology of cell death in glaucoma. Eye 2007, 21 (Suppl. 1), S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183–2193. [Google Scholar] [CrossRef]
- Rohrer, B.; Korenbrot, J.I.; LaVail, M.M.; Reichardt, L.F.; Xu, B. Role of neurotrophin receptor TrkB in the maturation of rod photoreceptors and establishment of synaptic transmission to the inner retina. J. Neurosci. 1999, 19, 8919–8930. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Harada, C.; Nakayama, N.; Okuyama, S.; Yoshida, K.; Kohsaka, S.; Matsuda, H.; Wada, K. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron 2000, 26, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Wahlin, K.J.; Campochiaro, P.A.; Zack, D.J.; Adler, R. Neurotrophic factors cause activation of intracellular signaling pathways in Muller cells and other cells of the inner retina, but not photoreceptors. Investig. Ophthalmol. Vis. Sci. 2000, 41, 927–936. [Google Scholar]
- Grishanin, R.N.; Yang, H.; Liu, X.; Donohue-Rolfe, K.; Nune, G.C.; Zang, K.; Xu, B.; Duncan, J.L.; Lavail, M.M.; Copenhagen, D.R.; et al. Retinal TrkB receptors regulate neural development in the inner, but not outer, retina. Mol. Cell. Neurosci. 2008, 38, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.; Fong, A.L.; Dwyer, J.M.; Davare, M.; Reese, E.; Obrietan, K.; Impey, S. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J. Neurosci. 2004, 24, 4324–4332. [Google Scholar] [CrossRef] [Green Version]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef]
- Braunger, B.M.; Ohlmann, A.; Koch, M.; Tanimoto, N.; Volz, C.; Yang, Y.; Bösl, M.R.; Cvekl, A.; Jägle, H.; Seeliger, M.W.; et al. Constitutive overexpression of Norrin activates Wnt/β-catenin and endothelin-2 signaling to protect photoreceptors from light damage. Neurobiol. Dis. 2013, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Quigley, H.; Anderson, D.R. The dynamics and location of axonal transport blockade by acute intraocular pressure elevation in primate optic nerve. Investig. Ophthalmol. Vis. Sci. 1976, 15, 606–616. [Google Scholar]
- Arango-Gonzalez, B.; Cellerino, A.; Kohler, K. Exogenous brain-derived neurotrophic factor (BDNF) reverts phenotypic changes in the retinas of transgenic mice lacking the BDNF gene. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, T.; Bahring, R.; Carroll, P.; Grantyn, R. Repetitive firing deficits and reduced sodium current density in retinal ganglion cells developing in the absence of BDNF. J. Neurobiol. 1999, 40, 407–419. [Google Scholar] [CrossRef]
- Cellerino, A.; Carroll, P.; Thoenen, H.; Barde, Y.A. Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol. Cell. Neurosci. 1997, 9, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Zhu, L.; Lee, S.R.; Chung, S.H.; Gillies, M.C. Involvement of NT3 and P75(NTR) in photoreceptor degeneration following selective Muller cell ablation. J. Neuroinflamm. 2013, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Bai, Y.; Dergham, P.; Nedev, H.; Xu, J.; Galan, A.; Rivera, J.C.; ZhiHua, S.; Mehta, H.M.; Woo, S.B.; Sarunic, M.V.; et al. Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J. Biol. Chem. 2010, 285, 39392–39400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q. Actions of neurotrophic factors and their signaling pathways in neuronal survival and axonal regeneration. Mol. Neurobiol. 2006, 33, 155–179. [Google Scholar] [CrossRef]
- Frade, J.M.; Rodriguez-Tebar, A.; Barde, Y.A. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996, 383, 166–168. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, Growth Factors and BDNF-TrkB Signalling in Retinal Degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar] [CrossRef] [Green Version]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef]
- Bovolenta, P.; Frade, J.M.; Marti, E.; Rodriguez-Pena, M.A.; Barde, Y.A.; Rodriguez-Tebar, A. Neurotrophin-3 antibodies disrupt the normal development of the chick retina. J. Neurosci. 1996, 16, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Hayashi, K.; Stork, S.; Scheper, V.; Lenarz, T.; Forster, C.Y. The Conspicuous Link between Ear, Brain and Heart-Could Neurotrophin-Treatment of Age-Related Hearing Loss Help Prevent Alzheimer’s Disease and Associated Amyloid Cardiomyopathy? Biomolecules 2021, 11, 900. [Google Scholar] [CrossRef] [PubMed]
- Colafrancesco, V.; Parisi, V.; Sposato, V.; Rossi, S.; Russo, M.A.; Coassin, M.; Lambiase, A.; Aloe, L. Ocular application of nerve growth factor protects degenerating retinal ganglion cells in a rat model of glaucoma. J. Glaucoma 2011, 20, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; You, Y.; Li, J.; Gupta, V.; Golzan, M.; Klistorner, A.; van den Buuse, M.; Graham, S. BDNF impairment is associated with age-related changes in the inner retina and exacerbates experimental glaucoma. Biochim. Biophys. Acta 2014, 1842, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Lambiase, A.; Aloe, L.; Centofanti, M.; Parisi, V.; Bao, S.N.; Mantelli, F.; Colafrancesco, V.; Manni, G.L.; Bucci, M.G.; Bonini, S.; et al. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops: Implications for glaucoma. Proc. Natl. Acad. Sci. USA 2009, 106, 13469–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun-Julien, F.; Morquette, B.; Douillette, A.; Saragovi, H.U.; Di Polo, A. Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol. Cell. Neurosci. 2009, 40, 410–420. [Google Scholar] [CrossRef]
- Shi, Z.; Birman, E.; Saragovi, H.U. Neurotrophic rationale in glaucoma: A TrkA agonist, but not NGF or a p75 antagonist, protects retinal ganglion cells in vivo. Dev. Neurobiol. 2007, 67, 884–894. [Google Scholar] [CrossRef]
- Connor, T.B., Jr.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M.; et al. Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J. Clin. Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef]
- Pfeffer, B.A.; Flanders, K.C.; Guerin, C.J.; Danielpour, D.; Anderson, D.H. Transforming growth factor beta 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp. Eye Res. 1994, 59, 323–333. [Google Scholar] [CrossRef]
- Pasquale, L.R.; Dorman-Pease, M.E.; Lutty, G.A.; Quigley, H.A.; Jampel, H.D. Immunolocalization of TGF-beta 1, TGF-beta 2, and TGF-beta 3 in the anterior segment of the human eye. Investig. Ophthalmol. Vis. Sci. 1993, 34, 23–30. [Google Scholar]
- Nishida, K.; Sotozono, C.; Adachi, W.; Yamamoto, S.; Yokoi, N.; Kinoshita, S. Transforming growth factor-beta 1, -beta 2 and -beta 3 mRNA expression in human cornea. Curr. Eye Res. 1995, 14, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Guerin, C.J.; Hageman, G.S.; Pfeffer, B.A.; Flanders, K.C. Distribution of transforming growth factor-beta isoforms in the mammalian retina. J. Neurosci. Res. 1995, 42, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Lutty, G.A.; Merges, C.; Threlkeld, A.B.; Crone, S.; McLeod, D.S. Heterogeneity in localization of isoforms of TGF-beta in human retina, vitreous, and choroid. Investig. Ophthalmol. Vis. Sci. 1993, 34, 477–487. [Google Scholar]
- Hirsch, L.; Nazari, H.; Sreekumar, P.G.; Kannan, R.; Dustin, L.; Zhu, D.; Barron, E.; Hinton, D.R. TGF-beta2 secretion from RPE decreases with polarization and becomes apically oriented. Cytokine 2015, 71, 394–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvanta, A. Expression and secretion of transforming growth factor-beta in transformed and nontransformed retinal pigment epithelial cells. Ophthalmic Res. 1994, 26, 361–367. [Google Scholar] [CrossRef]
- Nagineni, C.N.; Cherukuri, K.S.; Kutty, V.; Detrick, B.; Hooks, J.J. Interferon-gamma differentially regulates TGF-beta1 and TGF-beta2 expression in human retinal pigment epithelial cells through JAK-STAT pathway. J. Cell. Physiol. 2007, 210, 192–200. [Google Scholar] [CrossRef]
- Bielmeier, C.B.; Roth, S.; Schmitt, S.I.; Boneva, S.K.; Schlecht, A.; Vallon, M.; Tamm, E.R.; Ergun, S.; Neueder, A.; Braunger, B.M. Transcriptional Profiling Identifies Upregulation of Neuroprotective Pathways in Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 6307. [Google Scholar] [CrossRef]
- Obata, H.; Kaji, Y.; Yamada, H.; Kato, M.; Tsuru, T.; Yamashita, H. Expression of transforming growth factor-beta superfamily receptors in rat eyes. Acta Ophthalmol. Scand. 1999, 77, 151–156. [Google Scholar] [CrossRef]
- Flanders, K.C. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004, 85, 47–64. [Google Scholar] [CrossRef]
- Kim, I.Y.; Kim, M.M.; Kim, S.J. Transforming growth factor-beta: Biology and clinical relevance. J. Biochem. Mol. Biol. 2005, 38, 1–8. [Google Scholar] [CrossRef]
- Klenkler, B.; Sheardown, H. Growth factors in the anterior segment: Role in tissue maintenance, wound healing and ocular pathology. Exp. Eye Res. 2004, 79, 677–688. [Google Scholar] [CrossRef]
- Schiller, M.; Javelaud, D.; Mauviel, A. TGF-beta-induced SMAD signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004, 35, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef] [Green Version]
- Saika, S.; Saika, S.; Liu, C.Y.; Azhar, M.; Sanford, L.P.; Doetschman, T.; Gendron, R.L.; Kao, C.W.; Kao, W.W. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev. Biol. 2001, 240, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Allinson, K.R.; Lee, H.S.; Fruttiger, M.; McCarty, J.H.; Arthur, H.M. Endothelial expression of TGFbeta type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS ONE 2012, 7, e39336. [Google Scholar] [CrossRef]
- Goumans, M.J.; Zwijsen, A.; van Rooijen, M.A.; Huylebroeck, D.; Roelen, B.A.; Mummery, C.L. Transforming growth factor-beta signalling in extraembryonic mesoderm is required for yolk sac vasculogenesis in mice. Development 1999, 126, 3473–3483. [Google Scholar] [CrossRef]
- Siqueira, M.; Francis, D.; Gisbert, D.; Gomes, F.C.A.; Stipursky, J. Radial Glia Cells Control Angiogenesis in the Developing Cerebral Cortex Through TGF-beta1 Signaling. Mol. Neurobiol. 2018, 55, 3660–3675. [Google Scholar] [CrossRef]
- Braunger, B.M.; Fuchshofer, R.; Tamm, E.R. The aqueous humor outflow pathways in glaucoma: A unifying concept of disease mechanisms and causative treatment. Eur. J. Pharm. Biopharm. 2015, 95, 173–181. [Google Scholar] [CrossRef]
- Fuchshofer, R. The pathogenic role of transforming growth factor-β2 in glaucomatous damage to the optic nerve head. Exp. Eye Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Munoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P.; Eye Diseases Prevalence Research Group. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Guyer, D.R.; Fine, S.L.; Maguire, M.G.; Hawkins, B.S.; Owens, S.L.; Murphy, R.P. Subfoveal choroidal neovascular membranes in age-related macular degeneration. Visual prognosis in eyes with relatively good initial visual acuity. Arch. Ophthalmol. 1986, 104, 702–705. [Google Scholar] [CrossRef]
- Wong, T.Y.; Chakravarthy, U.; Klein, R.; Mitchell, P.; Zlateva, G.; Buggage, R.; Fahrbach, K.; Probst, C.; Sledge, I. The natural history and prognosis of neovascular age-related macular degeneration: A systematic review of the literature and meta-analysis. Ophthalmology 2008, 115, 116–126. [Google Scholar] [CrossRef]
- Holz, F.G.; Strauss, E.C.; Schmitz-Valckenberg, S.; van Lookeren Campagne, M. Geographic atrophy: Clinical features and potential therapeutic approaches. Ophthalmology 2014, 121, 1079–1091. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog. Retin. Eye Res. 2015, 49, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Mammadzada, P.; Corredoira, P.M.; Andre, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell. Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlingemann, R.O. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 91–101. [Google Scholar] [CrossRef]
- Sulzbacher, F.; Pollreisz, A.; Kaider, A.; Kickinger, S.; Sacu, S.; Schmidt-Erfurth, U.; Vienna Eye Study Center. Identification and clinical role of choroidal neovascularization characteristics based on optical coherence tomography angiography. Acta Ophthalmol. 2017, 95, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Funk, M.; Karl, D.; Georgopoulos, M.; Benesch, T.; Sacu, S.; Polak, K.; Zlabinger, G.J.; Schmidt-Erfurth, U. Neovascular age-related macular degeneration: Intraocular cytokines and growth factors and the influence of therapy with ranibizumab. Ophthalmology 2009, 116, 2393–2399. [Google Scholar] [CrossRef]
- Wecker, T.; Ehlken, C.; Buhler, A.; Lange, C.; Agostini, H.; Bohringer, D.; Stahl, A. Five-year visual acuity outcomes and injection patterns in patients with pro-re-nata treatments for AMD, DME, RVO and myopic CNV. Br. J. Ophthalmol. 2017, 101, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulaiman, R.S.; Quigley, J.; Qi, X.; O’Hare, M.N.; Grant, M.B.; Boulton, M.E.; Corson, T.W. A Simple Optical Coherence Tomography Quantification Method for Choroidal Neovascularization. J. Ocul. Pharmacol. Ther. 2015, 31, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, V.; Lecomte, J.; Hansen, S.; Blacher, S.; Gonzalez, M.L.; Struman, I.; Sounni, N.E.; Rozet, E.; de Tullio, P.; Foidart, J.M.; et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc. 2013, 8, 2197–2211. [Google Scholar] [CrossRef]
- Lois, N.; McCarter, R.V.; O’Neill, C.; Medina, R.J.; Stitt, A.W. Endothelial progenitor cells in diabetic retinopathy. Front. Endocrinol. 2014, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vis. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simo, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Mey, J.; Thanos, S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993, 602, 304–317. [Google Scholar] [CrossRef]
- Mysona, B.A.; Shanab, A.Y.; Elshaer, S.L.; El-Remessy, A.B. Nerve growth factor in diabetic retinopathy: Beyond neurons. Expert Rev. Ophthalmol. 2014, 9, 99–107. [Google Scholar] [CrossRef]
- Afarid, M.; Torabi-Nami, M.; Nemati, A.; Khosravi, A.; Malekzadeh, M. Brain-derived neurotrophic factor in patients with advanced age-related macular degeneration. Int. J. Ophthalmol. 2015, 8, 991–995. [Google Scholar] [CrossRef]
- Cristofaro, B.; Stone, O.A.; Caporali, A.; Dawbarn, D.; Ieronimakis, N.; Reyes, M.; Madeddu, P.; Bates, D.O.; Emanueli, C. Neurotrophin-3 is a novel angiogenic factor capable of therapeutic neovascularization in a mouse model of limb ischemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1143–1150. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Tan, F.; Li, Z.; Thiele, C.J. NGF activation of TrkA induces vascular endothelial growth factor expression via induction of hypoxia-inducible factor-1alpha. Mol. Cell. Neurosci. 2011, 46, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Nico, B.; Mangieri, D.; Benagiano, V.; Crivellato, E.; Ribatti, D. Nerve growth factor as an angiogenic factor. Microvasc. Res. 2008, 75, 135–141. [Google Scholar] [CrossRef]
- Jee, D.; Lee, W.K. Inhibitory effect of intravitreal injection of bevacizumab on nerve growth factor. Curr. Eye Res. 2012, 37, 408–415. [Google Scholar] [CrossRef]
- Steinle, J.J.; Granger, H.J. Nerve growth factor regulates human choroidal, but not retinal, endothelial cell migration and proliferation. Auton. Neurosci. 2003, 108, 57–62. [Google Scholar] [CrossRef]
- Inanc Tekin, M.; Sekeroglu, M.A.; Demirtas, C.; Tekin, K.; Doguizi, S.; Bayraktar, S.; Yilmazbas, P. Brain-Derived Neurotrophic Factor in Patients With Age-Related Macular Degeneration and Its Correlation With Retinal Layer Thicknesses. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2833–2840. [Google Scholar] [CrossRef] [Green Version]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients With Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5594–5603. [Google Scholar] [CrossRef] [PubMed]
- Uzel, A.G.T.; UGurlu, N.; Toklu, Y.; CIcek, M.; Boral, B.; Sener, B.; CaGil, N. Relationship between Stages of Diabetic Retinopathy and Levels of Brain-Derived Neurotrophic Factor in Aqueous Humor and Serum. Retina 2020, 40, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Orlandini, M.; Galvagni, F. The Controversial Role of TGF-beta in Neovascular Age-Related Macular Degeneration Pathogenesis. Int. J. Mol. Sci. 2018, 19, 3363. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Liang, S.; Yu, W.; Zhao, M.; Huang, L.; Zhao, M.; Li, X. Semaphorin 3A blocks the formation of pathologic choroidal neovascularization induced by transforming growth factor beta. Mol. Vis. 2014, 20, 1258–1270. [Google Scholar] [PubMed]
- Ogata, N.; Yamamoto, C.; Miyashiro, M.; Yamada, H.; Matsushima, M.; Uyama, M. Expression of transforming growth factor-beta mRNA in experimental choroidal neovascularization. Curr. Eye Res. 1997, 16, 9–18. [Google Scholar] [CrossRef]
- Wang, X.; Ma, W.; Han, S.; Meng, Z.; Zhao, L.; Yin, Y.; Wang, Y.; Li, J. TGF-beta participates choroid neovascularization through Smad2/3-VEGF/TNF-alpha signaling in mice with Laser-induced wet age-related macular degeneration. Sci. Rep. 2017, 7, 9672. [Google Scholar] [CrossRef] [Green Version]
- Recalde, S.; Zarranz-Ventura, J.; Fernandez-Robredo, P.; Garcia-Gomez, P.J.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-beta inhibition decreases diode laser-induced choroidal neovascularization development in rats: P17 and P144 peptides. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7090–7097. [Google Scholar] [CrossRef]
- Zarranz-Ventura, J.; Fernandez-Robredo, P.; Recalde, S.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-beta inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS ONE 2013, 8, e65434. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.M.; Neri, G.; Caldi, E.; Fusco, F.; Bacci, T.; Tarantello, A.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; et al. TGF-beta concentrations and activity are down-regulated in the aqueous humor of patients with neovascular age-related macular degeneration. Sci. Rep. 2018, 8, 8053. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Puklin, J.E.; Frank, R.N. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3178–3188. [Google Scholar]
- Kliffen, M.; Sharma, H.S.; Mooy, C.M.; Kerkvliet, S.; de Jong, P.T. Increased expression of angiogenic growth factors in age-related maculopathy. Br. J. Ophthalmol. 1997, 81, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yafai, Y.; Iandiev, I.; Lange, J.; Unterlauft, J.D.; Wiedemann, P.; Bringmann, A.; Reichenbach, A.; Eichler, W. Muller glial cells inhibit proliferation of retinal endothelial cells via TGF-beta2 and Smad signaling. Glia 2014, 62, 1476–1485. [Google Scholar] [CrossRef]
- Schlecht, A.; Boneva, S.; Gruber, M.; Zhang, P.; Horres, R.; Bucher, F.; Auw-Haedrich, C.; Hansen, L.; Stahl, A.; Hilgendorf, I.; et al. Transcriptomic Characterization of Human Choroidal Neovascular Membranes Identifies Calprotectin as a Novel Biomarker for Patients with Age-Related Macular Degeneration. Am. J. Pathol. 2020, 190, 1632–1642. [Google Scholar] [CrossRef]
- Skeie, J.M.; Mullins, R.F. Macrophages in neovascular age-related macular degeneration: Friends or foes? Eye 2009, 23, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Gerhardinger, C.; Dagher, Z.; Sebastiani, P.; Park, Y.S.; Lorenzi, M. The transforming growth factor-beta pathway is a common target of drugs that prevent experimental diabetic retinopathy. Diabetes 2009, 58, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Hirase, K.; Ikeda, T.; Sotozono, C.; Nishida, K.; Sawa, H.; Kinoshita, S. Transforming growth factor beta2 in the vitreous in proliferative diabetic retinopathy. Arch. Ophthalmol. 1998, 116, 738–741. [Google Scholar] [CrossRef] [Green Version]
- Beranek, M.; Kankova, K.; Benes, P.; Izakovicova-Holla, L.; Znojil, V.; Hajek, D.; Vlkova, E.; Vacha, J. Polymorphism R25P in the gene encoding transforming growth factor-beta (TGF-beta1) is a newly identified risk factor for proliferative diabetic retinopathy. Am. J. Med. Genet. 2002, 109, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Meyer-Schwickerath, R.; Klein, M.; Schatz, H.; Pfeiffer, A. Deficient activation and different expression of transforming growth factor-beta isoforms in active proliferative diabetic retinopathy and neovascular eye disease. Exp. Clin. Endocrinol. Diabetes 1999, 107, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Antonelli-Orlidge, A.; Saunders, K.B.; Smith, S.R.; D’Amore, P.A. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. USA 1989, 86, 4544–4548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Rifkin, D.B. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: Activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J. Cell Biol. 1989, 109, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Lee, N.Y. Emerging Roles of Transforming Growth Factor beta Signaling in Diabetic Retinopathy. J. Cell Physiol. 2017, 232, 486–489. [Google Scholar] [CrossRef]
- Buck, C.P.R.; Timothy, C.C.; Charles, R. CNS neurotrophins are biologically active and expressed by multiple cell types. J. Mol. Histol. 2021, 35, 771–783. [Google Scholar] [CrossRef]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Ruberti, F.; Capsoni, S.; Comparini, A.; Di Daniel, E.; Franzot, J.; Gonfloni, S.; Rossi, G.; Berardi, N.; Cattaneo, A. Phenotypic Knockout of Nerve Growth Factor in Adult Transgenic Mice Reveals Severe Deficits in Basal Forebrain Cholinergic Neurons, Cell Death in the Spleen, and Skeletal Muscle Dystrophy. J. Neurosci. 2000, 20, 2589–2601. [Google Scholar] [CrossRef]
- Ernfors, P.; Lee, K.-F.; Jaenisch, R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 1994, 368, 147–150. [Google Scholar] [CrossRef]
- Liu, X.; Ernfors, P.; Wu, H.; Jaenisch, R. Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature 1995, 375, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Tessarollo, L.; Tsoulfas, P.; Donovan, M.J.; Palko, M.E.; Blair-Flynn, J.; Hempstead, B.L.; Parada, L.F. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc. Natl. Acad. Sci. USA 1997, 94, 14776–14781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.F.; Li, E.; Huber, L.J.; Landis, S.C.; Sharpe, A.H.; Chao, M.V.; Jaenisch, R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 1992, 69, 737–749. [Google Scholar] [CrossRef]
- De Groot, C.J.A.; Montagne, L.; Barten, A.D.; Sminia, P.; Van Der Valk, P. Expression of Transforming Growth Factor (TGF)-β1, -β2, and -β3 Isoforms and TGF-β Type I and Type II Receptors in Multiple Sclerosis Lesions and Human Adult Astrocyte Cultures. J. Neuropathol. Exp. Neurol. 1999, 58, 174–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nataf, S.; Guillen, M.; Pays, L. TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands. Int. J. Mol. Sci. 2019, 20, 5934. [Google Scholar] [CrossRef] [Green Version]
- Meyers, E.A.; Kessler, J.A. TGF-β Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef] [Green Version]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.J.; Barnes, A.P.; Hand, R.; Polleux, F.; Ehlers, M.D. TGF-β Signaling Specifies Axons during Brain Development. Cell 2010, 142, 144–157. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.-L.; Lee, Y.J.; Shin, J.; Lee, E.; Park, S.O.; McCarty, J.H.; Oh, S.P. TGF-β signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab. Investig. 2011, 91, 1554–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, S.; Wurdak, H.; Ittner, L.M.; Ille, F.; Sumara, G.; Schmid, M.T.; Draganova, K.; Lang, K.S.; Paratore, C.; Leveen, P.; et al. Brain area-specific effect of TGF-beta signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2008, 2, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Hellbach, N.; Weise, S.C.; Vezzali, R.; Wahane, S.D.; Heidrich, S.; Roidl, D.; Pruszak, J.; Esser, J.S.; Vogel, T. Neural deletion of Tgfbr2 impairs angiogenesis through an altered secretome. Hum. Mol. Genet. 2014, 23, 6177–6190. [Google Scholar] [CrossRef]
- McCarty, J.H.; Lacy-Hulbert, A.; Charest, A.; Bronson, R.T.; Crowley, D.; Housman, D.; Savill, J.; Roes, J.R.; Hynes, R.O. Selective ablation of αv integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 2005, 132, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Proctor, J.M. Vascular Development of the Brain Requires 8 Integrin Expression in the Neuroepithelium. J. Neurosci. 2005, 25, 9940–9948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambier, S.; Gline, S.; Mu, D.; Collins, R.; Araya, J.; Dolganov, G.; Einheber, S.; Boudreau, N.; Nishimura, S.L. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: An angiogenic control switch. Am. J. Pathol. 2005, 166, 1883–1894. [Google Scholar] [CrossRef]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Chopp, M. Neurorestorative therapies for stroke: Underlying mechanisms and translation to the clinic. Lancet Neurol. 2009, 8, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Pareek, V.; Faiq, M.A.; Ghosh, S.K.; Kumari, C. ADULT NEUROGENESIS IN HUMANS: A Review of Basic Concepts, History, Current Research, and Clinical Implications. Innov. Clin. Neurosci. 2019, 16, 30–37. [Google Scholar]
- Ergul, A.; Alhusban, A.; Fagan, S.C. Angiogenesis. Stroke 2012, 43, 2270–2274. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Sobrino, M.; Rosell, A.; Hernandez-Guillamon, M.; Penalba, A.; Boada, C.; Domingues-Montanari, S.; Ribo, M.; Alvarez-Sabin, J.; Montaner, J. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis 2011, 216, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, Z.-B.; Zhuge, Q.; Zheng, W.; Shao, B.; Wang, B.; Sun, F.; Jin, K. Glial Scar Formation Occurs in the Human Brain after Ischemic Stroke. Int. J. Med. Sci. 2014, 11, 344–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicka, D.; Rogozinska, K.; Aleksy, M.; Witte, O.W.; Skangiel-Kramska, J. Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol. Exp. 2008, 68, 155–168. [Google Scholar]
- Lively, S.; Moxon-Emre, I.; Schlichter, L.C. SC1/Hevin and Reactive Gliosis After Transient Ischemic Stroke in Young and Aged Rats. J. Neuropathol. Exp. Neurol. 2011, 70, 913–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morioka, T.; Kalehua, A.N.; Streit, W.J. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 1993, 327, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Rolls, A.; Shechter, R.; Schwartz, M. The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 2009, 10, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Fan, G.; Hirt, L.; Fujii, M.; Matsushita, K.; Liu, X.; Jaenisch, R.; Moskowitz, M.A. Ischemic brain damage in mice after selectively modifying BDNF or NT4 gene expression. J. Cereb. Blood Flow Metab. 2000, 20, 139–144. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Kim, E.; Ratan, R.; Lee, F.S.; Cho, S. Genetic variant of BDNF (Val66Met) polymorphism attenuates stroke-induced angiogenic responses by enhancing anti-angiogenic mediator CD36 expression. J. Neurosci. 2011, 31, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Siironen, J.; Juvela, S.; Kanarek, K.; Vilkki, J.; Hernesniemi, J.; Lappalainen, J. The Met allele of the BDNF Val66Met polymorphism predicts poor outcome among survivors of aneurysmal subarachnoid hemorrhage. Stroke 2007, 38, 2858–2860. [Google Scholar] [CrossRef] [Green Version]
- Vilkki, J.; Lappalainen, J.; Juvela, S.; Kanarek, K.; Hernesniemi, J.A.; Siironen, J. Relationship of the Met allele of the brain-derived neurotrophic factor Val66Met polymorphism to memory after aneurysmal subarachnoid hemorrhage. Neurosurgery 2008, 63, 198–203; discussion 203. [Google Scholar] [CrossRef] [PubMed]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic Potential of Neurotrophins for Repair After Brain Injury: A Helping Hand From Biomaterials. Front. Neurosci. 2019, 13, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D. Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRX 2005, 2, 120–128. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Lee, D.C.; Chiu, I.M. Neural stem cells, neural progenitors, and neurotrophic factors. Cell Transpl. 2007, 16, 133–150. [Google Scholar] [CrossRef]
- Luan, X.; Qiu, H.; Hong, X.; Wu, C.; Zhao, K.; Chen, H.; Zhu, Z.; Li, X.; Shen, H.; He, J. High serum nerve growth factor concentrations are associated with good functional outcome at 3months following acute ischemic stroke. Clin. Chim. Acta 2019, 488, 20–24. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Overman, J.J.; Zhong, S.; Mueller, R.; Lynch, G.; Carmichael, S.T. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J. Neurosci. 2011, 31, 3766–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Cejudo, J.; Gutierrez-Fernandez, M.; Otero-Ortega, L.; Rodriguez-Frutos, B.; Fuentes, B.; Vallejo-Cremades, M.T.; Hernanz, T.N.; Cerdan, S.; Diez-Tejedor, E. Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 2015, 46, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zacharek, A.; Zhang, C.; Jiang, H.; Li, Y.; Roberts, C.; Lu, M.; Kapke, A.; Chopp, M. Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J. Neurosci. 2005, 25, 2366–2375. [Google Scholar] [CrossRef]
- Delivanoglou, N.; Boziki, M.; Theotokis, P.; Kesidou, E.; Touloumi, O.; Dafi, N.; Nousiopoulou, E.; Lagoudaki, R.; Grigoriadis, N.; Charalampopoulos, I.; et al. Spatio-temporal expression profile of NGF and the two-receptor system, TrkA and p75NTR, in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2020, 17, 41. [Google Scholar] [CrossRef]
- Oderfeld-Nowak, B.; Zaremba, M.; Micera, A.; Aloe, L. The upregulation of nerve growth factor receptors in reactive astrocytes of rat spinal cord during experimental autoimmune encephalomyelitis. Neurosci. Lett. 2001, 308, 165–168. [Google Scholar] [CrossRef]
- Soilu-Hanninen, M.; Epa, R.; Shipham, K.; Butzkueven, H.; Bucci, T.; Barrett, G.; Bartlett, P.F.; Kilpatrick, T.J. Treatment of experimental autoimmune encephalomyelitis with antisense oligonucleotides against the low affinity neurotrophin receptor. J. Neurosci. Res. 2000, 59, 712–721. [Google Scholar] [CrossRef]
- Nataf, S.; Naveilhan, P.; Sindji, L.; Darcy, F.; Brachet, P.; Montero-Menei, C.N. Low affinity NGF receptor expression in the central nervous system during experimental allergic encephalomyelitis. J. Neurosci. Res. 1998, 52, 83–92. [Google Scholar] [CrossRef]
- Simone, D.R.; Micera, A.; Tirassa, P.; Aloe, L. mRNA for NGF and p75 in the central nervous system of rats affected by experimental allergic encephalomyelitis. Neuropathol. Appl. Neurobiol. 1996, 22, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Copray, S.; Küst, B.; Emmer, B.; Young Lin, M.; Liem, R.; Amor, S.; De Vries, H.; Floris, S.; Boddeke, E. Deficient p75 low-affinity neurotrophin receptor expression exacerbates experimental allergic encephalomyelitis in C57/BL6 mice. J. Neuroimmunol. 2004, 148, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Boutin, H.; Lefeuvre, R.A.; Horai, R.; Asano, M.; Iwakura, Y.; Rothwell, N.J. Role of IL-1α and IL-1β in Ischemic Brain Damage. J. Neurosci. 2001, 21, 5528–5534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Chinookoswong, N.; Miller, G. The Interleukin-1 Receptor Antagonist (rhIL-1ra) Protects against Cerebral Infarction in a Rat Model of Hypoxia-Ischemia. Exp. Neurol. 1994, 130, 362–367. [Google Scholar] [CrossRef]
- Sun, M.; Deng, B.; Zhao, X.; Gao, C.; Yang, L.; Zhao, H.; Yu, D.; Zhang, F.; Xu, L.; Chen, L.; et al. Isoflurane preconditioning provides neuroprotection against stroke by regulating the expression of the TLR4 signalling pathway to alleviate microglial activation. Sci. Rep. 2015, 5, 11445. [Google Scholar] [CrossRef]
- Yuan, Y.; Zha, H.; Rangarajan, P.; Ling, E.-A.; Wu, C. Anti-inflammatory effects of Edaravone and Scutellarin in activated microglia in experimentally induced ischemia injury in rats and in BV-2 microglia. BMC Neurosci. 2014, 15, 125. [Google Scholar] [CrossRef] [Green Version]
- Lavine, S.D.; Hofman, F.M.; Zlokovic, B.V. Circulating Antibody against Tumor Necrosis Factor–Alpha Protects Rat Brain from Reperfusion Injury. J. Cereb. Blood Flow Metab. 1998, 18, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.J. Targeting the central nervous system inflammatory response in ischemic stroke. Curr. Opin. Neurol. 2001, 14, 349–353. [Google Scholar] [CrossRef]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Disord. 2018, 11, 175628641878985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, H.Y.F.; Rawji, K.S.; Ghorbani, S.; Xue, M.; Yong, V.W. The benefits of neuroinflammation for the repair of the injured central nervous system. Cell. Mol. Immunol. 2019, 16, 540–546. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Neuroinflammation in Post-Ischemic Neurodegeneration of the Brain: Friend, Foe, or Both? Int. J. Mol. Sci. 2021, 22, 4405. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuker, C.; Strecker, J.-K.; Rawal, R.; Schmidt-Pogoda, A.; Ruck, T.; Wiendl, H.; Klotz, L.; Schäbitz, W.-R.; Sommer, C.J.; Minnerup, H.; et al. Immune Cell Infiltration into the Brain After Ischemic Stroke in Humans Compared to Mice and Rats: A Systematic Review and Meta-Analysis. Transl. Stroke Res. 2021, 11. [Google Scholar] [CrossRef]
- Dobolyi, A.; Vincze, C.; Pál, G.; Lovas, G. The Neuroprotective Functions of Transforming Growth Factor Beta Proteins. Int. J. Mol. Sci. 2012, 13, 8219–8258. [Google Scholar] [CrossRef] [PubMed]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFbeta signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Zhu, H.; Gui, Q.; Hui, X.; Wang, X.; Jiang, J.; Ding, L.; Sun, X.; Wang, Y.; Chen, H. TGF-beta1/Smad3 Signaling Pathway Suppresses Cell Apoptosis in Cerebral Ischemic Stroke Rats. Med. Sci. Monit. 2017, 23, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich-Noack, P.; Prehn, J.H.; Krieglstein, J. TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms. Stroke 1996, 27, 1609–1614; discussion 1615. [Google Scholar] [CrossRef]
- McNeill, H.; Williams, C.; Guan, J.; Dragunow, M.; Lawlor, P.; Sirimanne, E.; Nikolics, K.; Gluckman, P. Neuronal rescue with transforming growth factor-beta 1 after hypoxic-ischaemic brain injury. Neuroreport 1994, 5, 901–904. [Google Scholar] [CrossRef]
- Sugimoto, K.; Nishioka, R.; Ikeda, A.; Mise, A.; Takahashi, H.; Yano, H.; Kumon, Y.; Ohnishi, T.; Tanaka, J. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia 2014, 62, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liu, F.-F.; Liu, C.-Y.; Li, X.-P.; Zheng, S.-Z.; Li, Q.-Q.; Liu, Q. Neuroprotective effects of SMADs in a rat model of cerebral ischemia/reperfusion. Neural Regen. Res. 2015, 10, 438. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Fathali, N.; Doyle, K.P.; Williams, A.M.; Han, J.; Buckwalter, M.S. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 2014, 62, 1227–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollander, M.C.; Latour, L.L.; Yang, D.; Ishii, H.; Xiao, Z.; Min, Y.; Ray-Choudhury, A.; Munasinghe, J.; Merchant, A.S.; Lin, P.C.; et al. Attenuation of Myeloid-Specific TGFβ Signaling Induces Inflammatory Cerebrovascular Disease and Stroke. Circ. Res. 2017, 121, 1360–1369. [Google Scholar] [CrossRef]
- Sie, M.P.; Uitterlinden, A.G.; Bos, M.J.; Arp, P.P.; Breteler, M.M.; Koudstaal, P.J.; Pols, H.A.; Hofman, A.; van Duijn, C.M.; Witteman, J.C. TGF-beta 1 polymorphisms and risk of myocardial infarction and stroke: The Rotterdam Study. Stroke 2006, 37, 2667–2671. [Google Scholar] [CrossRef] [PubMed]
- Dunning, A.M.; Ellis, P.D.; McBride, S.; Kirschenlohr, H.L.; Healey, C.S.; Kemp, P.R.; Luben, R.N.; Chang-Claude, J.; Mannermaa, A.; Kataja, V.; et al. A transforming growth factorbeta1 signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer Res. 2003, 63, 2610–2615. [Google Scholar]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef]
- Prigoda, N.L.; Savas, S.; Abdalla, S.A.; Piovesan, B.; Rushlow, D.; Vandezande, K.; Zhang, E.; Ozcelik, H.; Gallie, B.L.; Letarte, M. Hereditary haemorrhagic telangiectasia: Mutation detection, test sensitivity and novel mutations. J. Med. Genet. 2006, 43, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Saad, B.; Constam, D.B.; Ortmann, R.; Moos, M.; Fontana, A.; Schachner, M. Astrocyte-derived TGF-beta 2 and NGF differentially regulate neural recognition molecule expression by cultured astrocytes. J. Cell Biol. 1991, 115, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlecht, A.; Vallon, M.; Wagner, N.; Ergün, S.; Braunger, B.M. TGFβ-Neurotrophin Interactions in Heart, Retina, and Brain. Biomolecules 2021, 11, 1360. https://doi.org/10.3390/biom11091360
Schlecht A, Vallon M, Wagner N, Ergün S, Braunger BM. TGFβ-Neurotrophin Interactions in Heart, Retina, and Brain. Biomolecules. 2021; 11(9):1360. https://doi.org/10.3390/biom11091360
Chicago/Turabian StyleSchlecht, Anja, Mario Vallon, Nicole Wagner, Süleyman Ergün, and Barbara M. Braunger. 2021. "TGFβ-Neurotrophin Interactions in Heart, Retina, and Brain" Biomolecules 11, no. 9: 1360. https://doi.org/10.3390/biom11091360
APA StyleSchlecht, A., Vallon, M., Wagner, N., Ergün, S., & Braunger, B. M. (2021). TGFβ-Neurotrophin Interactions in Heart, Retina, and Brain. Biomolecules, 11(9), 1360. https://doi.org/10.3390/biom11091360