
Mass Spectrometry-Based Structural Proteomics for Metal Ion/Protein Binding Studies


Abstract
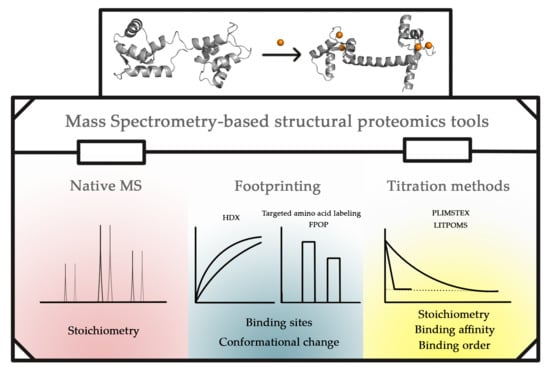
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MS Tools for Structural Proteomics and Metal Ion Binding
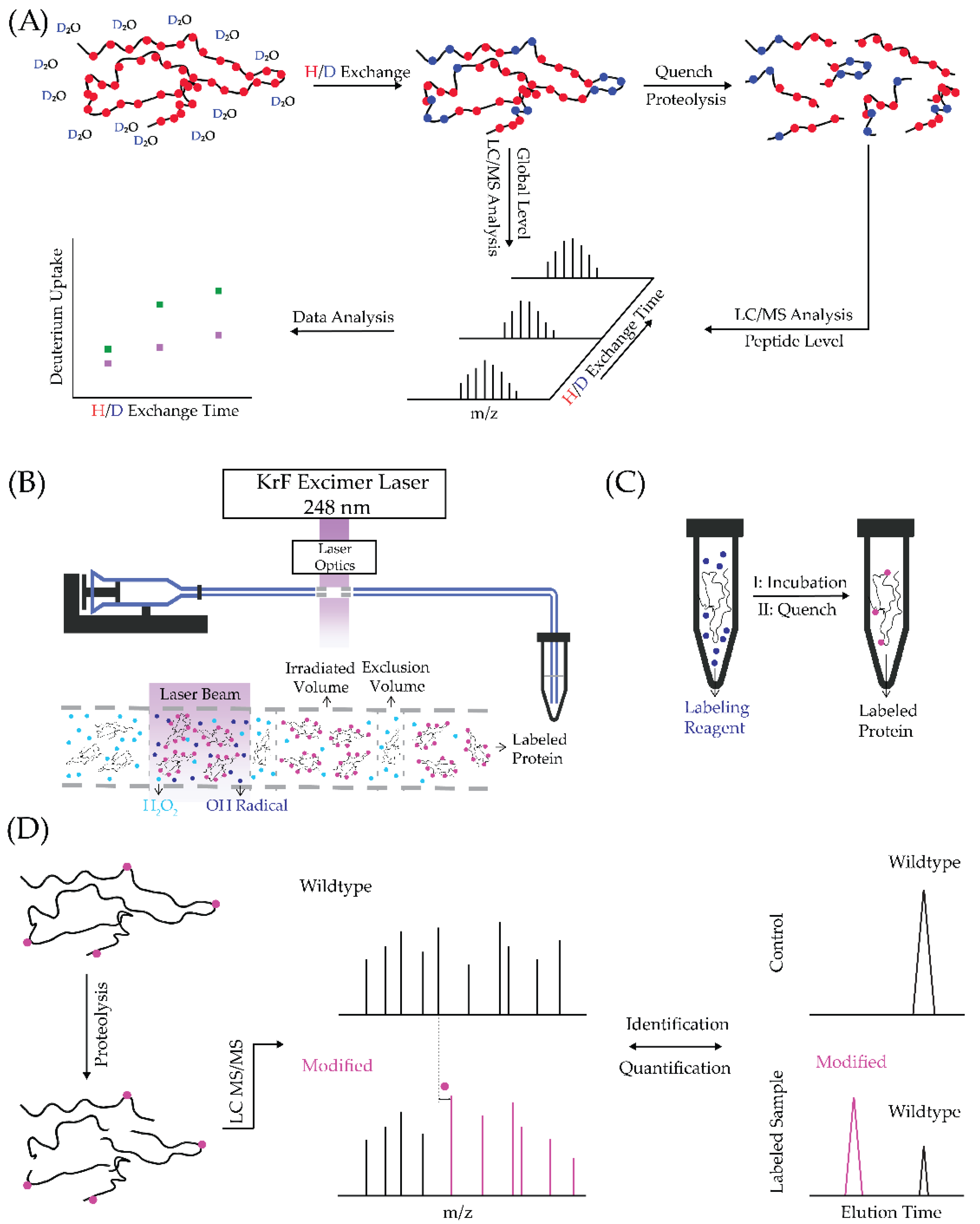
2.1. Hydrogen/Deuterium Exchange MS (HDX-MS)
2.2. Fast Photochemical Oxidation of Proteins (FPOP)
2.3. Specific Amino-Acid Footprinting
2.4. Native MS
3. Qualitative Studies and Stoichiometry Determination by MS-Based Methods
3.1. Determining Stoichiometry by Native MS
3.1.1. Mn2+ as Co-Factor for SFTSV Endonuclease
3.1.2. Iron Binding by Sidercalin
3.1.3. Ca2+ Binding to Centrin
3.2. IM-MS for Qualitative Studies of Ca2+ Binding to Calmodulin
3.3. Native Top-Down MS for Study of Metal Ions Binding to Aβ42
3.4. Titration Methods for Stoichiometry and Affinity
4. Identification of Metal Binding Site and Resulting Conformational Changes
4.1. HDX-MS
4.1.1. Ca2+ Binding to Calmodulin
4.1.2. Ca2+ Binding to DREAM
4.1.3. Ca2+ Binding to Human Centrin 2
4.1.4. Zinc Ion Binding to Hepatitis B Virus X
4.1.5. Iron Binding by Sidercalin
4.1.6. Interaction between Cu2+ and Aβ42
4.2. Targeted AminoAcid Footprinting
4.2.1. Iron Binding by Sidercalin
4.2.2. Zinc Ion Binding to Hepatitis B Virus X
4.2.3. Benzhydrazide Targeting Glu and Asp: Metal Ion Binding to Calmodulin
4.2.4. DEPC Footprinting of Cu2+ and Zn2+ Binding Sites
4.2.5. Cross-Linking: Ca2+ Mediated Calmodulin–bMunc13-2 Interaction
4.3. Hydroxyl Radical Footprinting of Calmodulin
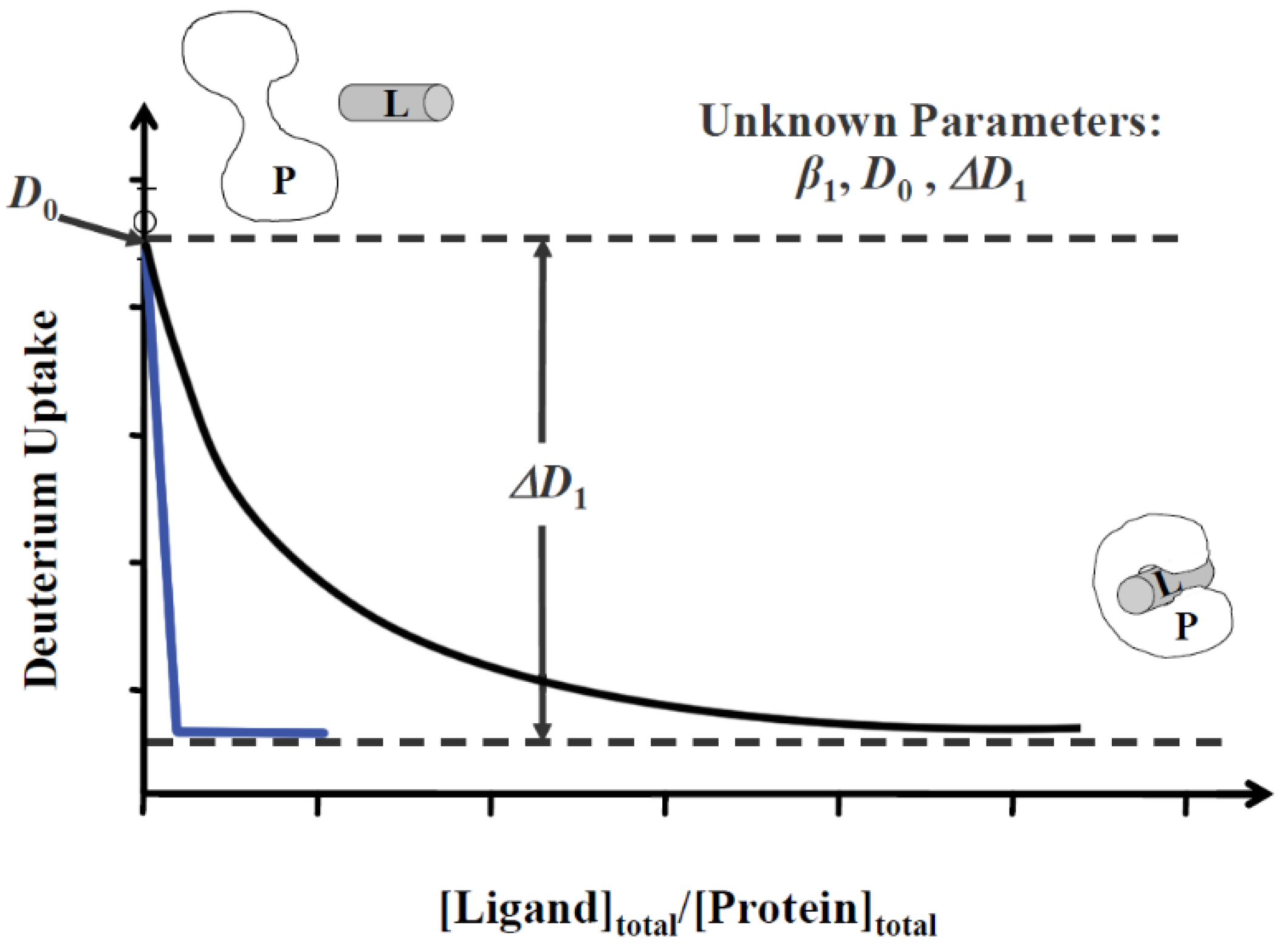
5. Determining Affinity and Binding Order by MS Titration Methods
5.1. Protein–Ligand Interactions in Solution by Mass Spectrometry, Titration and HDX Exchange (PLIMSTEX)
5.2. Protein–Ligand Interaction by Ligand Titration, Fast Photochemical Oxidation of Proteins and Mass Spectrometry (LITPOMS)
6. Illustration of Integrated Methods: Ca2+ Binding to Calprotectin
7. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zambelli, B.; Musiani, F.; Ciurli, S. Metal ion-mediated DNA-protein interactions. Met. Ions Life Sci. 2012, 10, 135–170. [Google Scholar]
- Rosenzweig, A.C. Metallochaperones. Chem. Biol. 2002, 9, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Perozzo, R.; Folkers, G.; Scapozza, L. Thermodynamics of protein-ligand interactions: History, presence, and future aspects. J. Recept. Signal Transduct. Res. 2004, 24, 1–52. [Google Scholar] [CrossRef]
- Concepcion, J.; Witte, K.; Wartchow, C.; Choo, S.; Yao, D.F.; Persson, H.; Wei, J.; Li, P.; Heidecker, B.; Ma, W.L.; et al. Label-Free Detection of Biomolecular Interactions Using BioLayer Interferometry for Kinetic Characterization. Comb. Chem. High Throughput Screen. 2009, 12, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Torreri, P.; Ceccarini, M.; Macioce, P.; Petrucci, T.C. Biomolecular interactions by Surface Plasmon Resonance technology. Ann. Ist. Super. Sanita 2005, 41, 437–441. [Google Scholar]
- Rossi, A.M.; Taylor, C.W. Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc. 2011, 6, 365–387. [Google Scholar] [CrossRef] [Green Version]
- Ranjbar, B.; Gill, P. Circular Dichroism Techniques: Biomolecular and Nanostructural Analyses—A Review. Chem. Biol. Drug Des. 2009, 74, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y. Single-particle cryo-EM—How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orts, J.; Gossert, A.D. Structure determination of protein-ligand complexes by NMR in solution. Methods 2018, 138–139, 3–25. [Google Scholar] [CrossRef]
- Shi, Y. A Glimpse of Structural Biology through X-ray Crystallography. Cell 2014, 159, 995–1014. [Google Scholar] [CrossRef] [Green Version]
- Sheshberadaran, H.; Payne, L.G. Protein antigen-monoclonal antibody contact sites investigated by limited proteolysis of monoclonal antibody-bound antigen: Protein “footprinting”. Proc. Natl. Acad. Sci. USA 1988, 85, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.R.; Zhang, M.M.; Gross, M.L. Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem. Rev. 2020, 120, 4355–4454. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [Green Version]
- James, E.I.; Murphree, T.A.; Vorauer, C.; Engen, J.R.; Guttman, M. Advances in Hydrogen/Deuterium Exchange Mass Spectrometry and the Pursuit of Challenging Biological Systems. Chem. Rev. 2021. [Google Scholar] [CrossRef]
- Walters, B.T.; Ricciuti, A.; Mayne, L.; Englander, S.W. Minimizing Back Exchange in the Hydrogen Exchange-Mass Spectrometry Experiment. J. Am. Soc. Mass Spectrom. 2012, 23, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Cravello, L.; Lascoux, D.; Forest, E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. 2003, 17, 2387–2393. [Google Scholar] [CrossRef]
- Hambly, D.M.; Gross, M.L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chance, M.R. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, G.; Wei, H.; Huang, R.Y.-C.; Mo, J.; Rempel, D.L.; Tymiak, A.A.; Gross, M.L. Fast Photochemical Oxidation of Proteins (FPOP) Maps the Epitope of EGFR Binding to Adnectin. J. Am. Soc. Mass Spectrom. 2014, 25, 2084–2092. [Google Scholar] [CrossRef] [Green Version]
- Li, K.S.; Shi, L.; Gross, M.L. Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization. Acc. Chem. Res. 2018, 51, 736–744. [Google Scholar] [CrossRef]
- Vahidi, S.; Konermann, L. Probing the Time Scale of FPOP (Fast Photochemical Oxidation of Proteins): Radical Reactions Extend Over Tens of Milliseconds. J. Am. Soc. Mass Spectrom. 2016, 27, 1156–1164. [Google Scholar] [CrossRef]
- Liu, X.R.; Rempel, D.L.; Gross, M.L. Protein higher-order-structure determination by fast photochemical oxidation of proteins and mass spectrometry analysis. Nat. Protoc. 2020, 15, 3942–3970. [Google Scholar] [CrossRef]
- Gregory, D.S.; Martin, A.C.R.; Cheetham, J.C.; Rees, A.R. The prediction and characterization of metal binding sites in proteins. Protein Eng. Des. Sel. 1993, 6, 29–35. [Google Scholar] [CrossRef]
- Mendoza, V.L.; Vachet, R.W. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 2009, 28, 785–815. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lantz, C.; Brown, K.A.; Ge, Y.; Paša-Tolić, L.; Loo, J.A.; Lermyte, F. Higher-order structural characterisation of native proteins and complexes by top-down mass spectrometry. Chem. Sci. 2020, 11, 12918–12936. [Google Scholar] [CrossRef]
- Leney, A.C.; Heck, A.J.R. Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Shin, W.-J.; Zhang, B.; Choi, Y.; Yoo, J.-S.; Zimmerman, M.I.; Frederick, T.E.; Bowman, G.R.; Gross, M.L.; Leung, D.W.; et al. The Cap-Snatching SFTSV Endonuclease Domain Is an Antiviral Target. Cell Rep. 2020, 30, 153–163.e5. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012, 10, 51–65. [Google Scholar] [CrossRef]
- Fernández-García, Y.; Reguera, J.; Busch, C.; Witte, G.; Sánchez-Ramos, O.; Betzel, C.; Cusack, S.; Günther, S.; Reindl, S. Atomic Structure and Biochemical Characterization of an RNA Endonuclease in the N Terminus of Andes Virus L Protein. PLoS Pathog. 2016, 12, e1005635. [Google Scholar] [CrossRef]
- Doan, L.; Handa, B.; Roberts, N.A.; Klumpp, K. Metal Ion Catalysis of RNA Cleavage by the Influenza Virus Endonuclease. Biochemistry 1999, 38, 5612–5619. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.; Kopicki, J.-D.; Busch, C.; Olschewski, S.; Rosenthal, M.; Uetrecht, C.; Günther, S.; Reindl, S. Biochemical and structural studies reveal differences and commonalities among cap-snatching endonucleases from segmented negative-strand RNA viruses. J. Biol. Chem. 2018, 293, 19686–19698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abergel, R.J.; Clifton, M.C.; Pizarro, J.C.; Warner, J.A.; Shuh, D.K.; Strong, R.K.; Raymond, K.N. The Siderocalin/Enterobactin Interaction: A Link between Mammalian Immunity and Bacterial Iron Transport1. J. Am. Chem. Soc. 2008, 130, 11524–11534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, M.C.; Rupert, P.B.; Hoette, T.M.; Raymond, K.N.; Abergel, R.J.; Strong, R.K. Parsing the functional specificity of Siderocalin/Lipocalin 2/NGAL for siderophores and related small-molecule ligands. J. Struct. Biol. X 2019, 2, 100008. [Google Scholar] [CrossRef]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef] [Green Version]
- Miethke, M.; Marahiel, M.A. Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef] [Green Version]
- Shields-Cutler, R.R.; Crowley, J.R.; Hung, C.S.; Stapleton, A.E.; Aldrich, C.C.; Marschall, J.; Henderson, J.P. Human Urinary Composition Controls Antibacterial Activity of Siderocalin. J. Biol. Chem. 2015, 290, 15949–15960. [Google Scholar] [CrossRef] [Green Version]
- Shields-Cutler, R.R.; Crowley, J.R.; Miller, C.D.; Stapleton, A.E.; Cui, W.; Henderson, J.P. Human Metabolome-derived Cofactors Are Required for the Antibacterial Activity of Siderocalin in Urine. J. Biol. Chem. 2016, 291, 25901–25910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Steinberg, L.K.; Cheng, M.; Song, J.H.; Henderson, J.P.; Gross, M.L. Site-Specific Siderocalin Binding to Ferric and Ferric-Free Enterobactin As Revealed by Mass Spectrometry. ACS Chem. Biol. 2020, 15, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.A.; Benson, L.M.; Bergen, H.R.; Venyaminov, S.Y.; Salisbury, J.L.; Ryan, Z.C.; Thompson, J.R.; Sperry, J.; Gross, M.L.; Kumar, R. Metal-binding properties of human centrin-2 determined by micro-electrospray ionization mass spectrometry and UV spectroscopy. J. Am. Soc. Mass Spectrom. 2006, 17, 1158–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salisbury, J.L.; Suino, K.M.; Busby, R.; Springett, M. Centrin-2 Is Required for Centriole Duplication in Mammalian Cells. Curr. Biol. 2002, 12, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Dantas, T.J.; Daly, O.M.; Conroy, P.C.; Tomas, M.; Wang, Y.; Lalor, P.; Dockery, P.; Ferrando-May, E.; Morrison, C.G. Calcium-Binding Capacity of Centrin2 Is Required for Linear POC5 Assembly but Not for Nucleotide Excision Repair. PLoS ONE 2013, 8, e68487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susa, A.C.; Xia, Z.; Williams, E.R. Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment. Anal. Chem. 2017, 89, 3116–3122. [Google Scholar] [CrossRef]
- Mortensen, D.N.; Williams, E.R. Surface-Induced Protein Unfolding in Submicron Electrospray Emitters. Anal. Chem. 2016, 88, 9662–9668. [Google Scholar] [CrossRef] [PubMed]
- Susa, A.C.; Xia, Z.; Williams, E.R. Native Mass Spectrometry from Common Buffers with Salts That Mimic the Extracellular Environment. Angew. Chem. Int. Ed. 2017, 56, 7912–7915. [Google Scholar] [CrossRef]
- Robinson, C.V.; Chung, E.W.; Kragelund, B.B.; Knudsen, J.; Aplin, R.T.; Poulsen, F.M.; Dobson, C.M. Probing the Nature of Noncovalent Interactions by Mass Spectrometry. A Study of Protein−CoA Ligand Binding and Assembly. J. Am. Chem. Soc. 1996, 118, 8646–8653. [Google Scholar] [CrossRef]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Uetrecht, C.; Rose, R.J.; Van Duijn, E.; Lorenzen, K.; Heck, A.J.R. Ion mobility mass spectrometry of proteins and proteinassemblies. Chem. Soc. Rev. 2010, 39, 1633–1655. [Google Scholar] [CrossRef] [PubMed]
- Mccabe, J.W.; Hebert, M.J.; Shirzadeh, M.; Mallis, C.S.; Denton, J.K.; Walker, T.E.; Russell, D.H. The Ims paradox: A perspective on structural ion mobility-mass spectrometry. Mass Spectrom. Rev. 2021, 40, 280–305. [Google Scholar] [CrossRef] [PubMed]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wongkongkathep, P.; Han, J.Y.; Choi, T.S.; Yin, S.; Kim, H.I.; Loo, J.A. Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am. Soc. Mass Spectrom. 2018, 29, 1870–1880. [Google Scholar] [CrossRef]
- Dong, S.; Shirzadeh, M.; Fan, L.; Laganowsky, A.; Russell, D.H. Ag+ Ion Binding to Human Metallothionein-2A Is Cooperative and Domain Specific. Anal. Chem. 2020, 92, 8923–8932. [Google Scholar] [CrossRef]
- Marcinko, T.M.; Liang, C.; Savinov, S.; Chen, J.; Vachet, R.W. Structural Heterogeneity in the Preamyloid Oligomers of β-2-Microglobulin. J. Mol. Biol. 2020, 432, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.-B. Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation. J. Inorg. Biochem. 2019, 194, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Faull, P.A.; Korkeila, K.E.; Kalapothakis, J.M.; Gray, A.; Mccullough, B.J.; Barran, P.E. Gas-phase metalloprotein complexes interrogated by ion mobility-mass spectrometry. Int. J. Mass Spectrom. 2009, 283, 140–148. [Google Scholar] [CrossRef]
- Kuboniwa, H.; Tjandra, N.; Grzesiek, S.; Ren, H.; Klee, C.B.; Bax, A. Solution structure of calcium-free calmodulin. Nat. Struct. Mol. Biol. 1995, 2, 768–776. [Google Scholar] [CrossRef]
- Wyttenbach, T.; Grabenauer, M.; Thalassinos, K.; Scrivens, J.H.; Bowers, M.T. The Effect of Calcium Ions and Peptide Ligands on the Relative Stabilities of the Calmodulin Dumbbell and Compact Structures. J. Phys. Chem. B 2010, 114, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cui, W.; Wen, J.; Blankenship, R.E.; Gross, M.L. Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal. Chem. 2011, 83, 5598–5606. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhang, J.; Yin, S.; Loo, J.A. Top-Down ESI-ECD-FT-ICR Mass Spectrometry Localizes Noncovalent Protein-Ligand Binding Sites. J. Am. Chem. Soc. 2006, 128, 14432–14433. [Google Scholar] [CrossRef]
- O’Brien, J.P.; Li, W.; Zhang, Y.; Brodbelt, J.S. Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2014, 136, 12920–12928. [Google Scholar] [CrossRef] [Green Version]
- Lermyte, F.; Everett, J.; Lam, Y.P.Y.; Wootton, C.A.; Brooks, J.; Barrow, M.P.; Telling, N.D.; Sadler, P.J.; O’Connor, P.B.; Collingwood, J.F. Metal Ion Binding to the Amyloid β Monomer Studied by Native Top-Down FTICR Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 2123–2134. [Google Scholar] [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S277–S281. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.M.; Rempel, D.L.; Du, Z.; Gross, M.L. Quantification of Protein−Ligand Interactions by Mass Spectrometry, Titration, and H/D Exchange: PLIMSTEX. J. Am. Chem. Soc. 2003, 125, 5252–5253. [Google Scholar] [CrossRef]
- Wiseman, T.; Williston, S.; Brandts, J.F.; Lin, L.-N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989, 179, 131–137. [Google Scholar] [CrossRef]
- Zhu, M.M.; Rempel, D.L.; Gross, M.L. Modeling data from titration, amide H/D exchange, and mass spectrometry to obtain protein-ligand binding constants. J. Am. Soc. Mass Spectrom. 2004, 15, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.M.; Chitta, R.; Gross, M.L. PLIMSTEX: A novel mass spectrometric method for the quantification of protein–ligand interactions in solution. Int. J. Mass Spectrom. 2005, 240, 213–220. [Google Scholar] [CrossRef]
- Liu, X.R.; Zhang, M.M.; Rempel, D.L.; Gross, M.L. Protein-Ligand Interaction by Ligand Titration, Fast Photochemical Oxidation of Proteins and Mass Spectrometry: LITPOMS. J. Am. Soc. Mass Spectrom. 2019, 30, 213–217. [Google Scholar] [CrossRef]
- Adhikari, J.; Stephan, J.R.; Rempel, D.L.; Nolan, E.M.; Gross, M.L. Calcium Binding to the Innate Immune Protein Human Calprotectin Revealed by Integrated Mass Spectrometry. J. Am. Chem. Soc. 2020, 142, 13372–13383. [Google Scholar] [CrossRef]
- Nemirovskiy, O.; Giblin, D.E.; Gross, M.L. Electrospray ionization mass spectrometry and hydrogen/deuterium exchange for probing the interaction of calmodulin with calcium. J. Am. Soc. Mass Spectrom. 1999, 10, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, J.; Craig, T.A.; Kumar, R.; Gross, M.L. Hydrogen–Deuterium Exchange Mass Spectrometry Reveals Calcium Binding Properties and Allosteric Regulation of Downstream Regulatory Element Antagonist Modulator (DREAM). Biochemistry 2017, 56, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Carrión, A.M.; Link, W.A.; Ledo, F.; Mellström, B.; Naranjo, J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.A.; Benson, L.M.; Venyaminov, S.Y.; Klimtchuk, E.S.; Bajzer, Z.; Prendergast, F.G.; Naylor, S.; Kumar, R. The Metal-binding Properties of DREAM. J. Biol. Chem. 2002, 277, 10955–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledo, F. Ca2+-dependent block of CREB-CBP transcription by repressor DREAM. EMBO J. 2002, 21, 4583–4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, M.; Tong, K.I.; Lilliehook, C.; Wasco, W.; Buxbaum, J.D.; Cheng, H.-Y.M.; Penninger, J.M.; Ikura, M.; Ames, J.B. Calcium-regulated DNA Binding and Oligomerization of the Neuronal Calcium-sensing Protein, Calsenilin/DREAM/KChIP3. J. Biol. Chem. 2001, 276, 41005–41013. [Google Scholar] [CrossRef] [Green Version]
- Carrión, A.M.; Mellstrom, B.; Naranjo, J.R. Protein Kinase A-Dependent Derepression of the Human Prodynorphin Gene via Differential Binding to an Intragenic Silencer Element. Mol. Cell. Biol. 1998, 18, 6921–6929. [Google Scholar] [CrossRef] [Green Version]
- Lusin, J.D.; Vanarotti, M.; Li, C.; Valiveti, A.; Ames, J.B. NMR Structure of DREAM: Implications for Ca2+-Dependent DNA Binding and Protein Dimerization. Biochemistry 2008, 47, 2252–2264. [Google Scholar] [CrossRef]
- Osawa, M.; Dace, A.; Tong, K.I.; Valiveti, A.; Ikura, M.; Ames, J.B. Mg2+ and Ca2+ Differentially Regulate DNA Binding and Dimerization of DREAM. J. Biol. Chem. 2005, 280, 18008–18014. [Google Scholar] [CrossRef] [Green Version]
- Sperry, J.B.; Ryan, Z.C.; Kumar, R.; Gross, M.L. Hydrogen/deuterium exchange reflects binding of human centrin 2 to Ca2+ and Xeroderma pigmentosum Group C peptide: An example of EX1 kinetics. Int. J. Mass Spectrom. 2012, 330–332, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Masutani, C.; Takemura, M.; Uchida, A.; Sugasawa, K.; Kondoh, J.; Ohkuma, Y.; Hanaoka, F. Centrosome Protein Centrin 2/Caltractin 1 is Part of the Xeroderma Pigmentosum Group C Complex that Initiates Global Genome Nucleotide Excision Repair. J. Biol. Chem. 2001, 276, 18665–18672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourbez, M.; Firanescu, C.; Yang, A.; Unipan, L.; Duchambon, P.; Blouquit, Y.; Craescu, C.T. Calcium-dependent Self-assembly of Human Centrin 2. J. Biol. Chem. 2004, 279, 47672–47680. [Google Scholar] [CrossRef] [Green Version]
- Durussel, I.; Blouquit, Y.; Middendorp, S.; Craescu, C.T.; Cox, J.A. Cation- and peptide-binding properties of human centrin 2. FEBS Lett. 2000, 472, 208–212. [Google Scholar] [CrossRef]
- Matei, E.; Miron, S.; Blouquit, Y.; Duchambon, P.; Durussel, I.; Cox, J.A.; Craescu, C.T. C-Terminal Half of Human Centrin 2 Behaves like a Regulatory EF-Hand Domain. Biochemistry 2003, 42, 1439–1450. [Google Scholar] [CrossRef]
- Ramakrishnan, D.; Xing, W.; Beran, R.K.; Chemuru, S.; Rohrs, H.; Niedziela-Majka, A.; Marchand, B.; Mehra, U.; Zábranský, A.; Doležal, M.; et al. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93, e00250-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [Green Version]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The Neutrophil Lipocalin NGAL Is a Bacteriostatic Agent that Interferes with Siderophore-Mediated Iron Acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Goetz, D.H.; Willie, S.T.; Armen, R.S.; Bratt, T.; Borregaard, N.; Strong, R.K. Ligand Preference Inferred from the Structure of Neutrophil Gelatinase Associated Lipocalin. Biochemistry 2000, 39, 1935–1941. [Google Scholar] [CrossRef]
- Pan, J.; Wilson, D.J.; Konermann, L. Pulsed Hydrogen Exchange and Electrospray Charge-State Distribution as Complementary Probes of Protein Structure in Kinetic Experiments: Implications for Ubiquitin Folding. Biochemistry 2005, 44, 8627–8633. [Google Scholar] [CrossRef]
- Roder, H.; Elöve, G.A.; Englander, S.W. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 1988, 335, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Rempel, D.L.; Zhang, J.; Sharma, A.K.; Mirica, L.M.; Gross, M.L. Pulsed hydrogen–deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Rob, T.; Liuni, P.; Gill, P.K.; Zhu, S.; Balachandran, N.; Berti, P.J.; Wilson, D.J. Measuring Dynamics in Weakly Structured Regions of Proteins Using Microfluidics-Enabled Subsecond H/D Exchange Mass Spectrometry. Anal. Chem. 2012, 84, 3771–3779. [Google Scholar] [CrossRef] [PubMed]
- Mitchell Wells, J.; Mcluckey, S.A. Collision-Induced Dissociation (CID) of Peptides and Proteins. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; pp. 148–185. [Google Scholar]
- Mistarz, U.H.; Bellina, B.; Jensen, P.F.; Brown, J.M.; Barran, P.E.; Rand, K.D. UV Photodissociation Mass Spectrometry Accurately Localize Sites of Backbone Deuteration in Peptides. Anal. Chem. 2018, 90, 1077–1080. [Google Scholar] [CrossRef] [Green Version]
- Rand, K.D.; Adams, C.M.; Zubarev, R.A.; Jørgensen, T.J.D. Electron Capture Dissociation Proceeds with a Low Degree of Intramolecular Migration of Peptide Amide Hydrogens. J. Am. Chem. Soc. 2008, 130, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Jørgensen, T.J.D. Development of a Peptide Probe for the Occurrence of Hydrogen (1H/2H) Scrambling upon Gas-Phase Fragmentation. Anal. Chem. 2007, 79, 8686–8693. [Google Scholar] [CrossRef]
- Zehl, M.; Rand, K.D.; Jensen, O.N.; Jørgensen, T.J.D. Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J. Am. Chem. Soc. 2008, 130, 17453–17459. [Google Scholar] [CrossRef]
- Chumsae, C.; Gifford, K.; Lian, W.; Liu, H.; Radziejewski, C.H.; Zhou, Z.S. Arginine Modifications by Methylglyoxal: Discovery in a Recombinant Monoclonal Antibody and Contribution to Acidic Species. Anal. Chem. 2013, 85, 11401–11409. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wang, Y. Site-Selective Modifications of Arginine Residues in Human Hemoglobin Induced by Methylglyoxal. Biochemistry 2006, 45, 15654–15660. [Google Scholar] [CrossRef]
- Makoff, A.J.; Malcolm, A.D.B. Properties of methyl acetimidate and its use as a protein-modifying reagent. Biochem. J. 1981, 193, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Gregory, J.D. The Stability of N-Ethylmaleimide and its Reaction with Sulfhydryl Groups. J. Am. Chem. Soc. 1955, 77, 3922–3923. [Google Scholar] [CrossRef]
- Leslie, J. Spectral shifts in the reaction of N-ethylmaleimide with proteins. Anal. Biochem. 1965, 10, 162–167. [Google Scholar] [CrossRef]
- Smyth, D.G.; Nagamatsu, A.; Fruton, J.S. Some Reactions of N-Ethylmaleimide1. J. Am. Chem. Soc. 1960, 82, 4600–4604. [Google Scholar] [CrossRef]
- Guo, C.; Cheng, M.; Gross, M.L. Protein-Metal-Ion Interactions Studied by Mass Spectrometry-Based Footprinting with Isotope-Encoded Benzhydrazide. Anal. Chem. 2019, 91, 1416–1423. [Google Scholar] [CrossRef]
- Gau, B.; Garai, K.; Frieden, C.; Gross, M.L. Mass Spectrometry-Based Protein Footprinting Characterizes the Structures of Oligomeric Apolipoprotein E2, E3, and E4. Biochemistry 2011, 50, 8117–8126. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Tomechko, S.E.; Kiselar, J.; Shi, W.; Deperalta, G.; Wecksler, A.T.; Gokulrangan, G.; Ling, V.; Chance, M.R. Characterizing monoclonal antibody structure by carboxyl group footprinting. mAbs 2015, 7, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Krawitz, D.; Callahan, M.D.; Deperalta, G.; Wecksler, A.T. Characterization of ELISA Antibody-Antigen Interaction using Footprinting-Mass Spectrometry and Negative Staining Transmission Electron Microscopy. J. Am. Soc. Mass Spectrom. 2018, 29, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Wecksler, A.T.; Kalo, M.S.; Deperalta, G. Mapping of Fab-1:VEGF Interface Using Carboxyl Group Footprinting Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 2077–2080. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, H.; Gross, M.L.; Blankenship, R.E. Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc. Natl. Acad. Sci. USA 2009, 106, 6134–6139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shen, W.; Rempel, D.; Monsey, J.; Vidavsky, I.; Gross, M.L.; Bose, R. Carboxyl-Group Footprinting Maps the Dimerization Interface and Phosphorylation-induced Conformational Changes of a Membrane-associated Tyrosine Kinase. Mol. Cell. Proteom. 2011, 10, M110.005678. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Senguen, F.T.; Grabarek, Z. X-ray Structures of Magnesium and Manganese Complexes with the N-Terminal Domain of Calmodulin: Insights into the Mechanism and Specificity of Metal Ion Binding to an EF-Hand. Biochemistry 2012, 51, 6182–6194. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, V.L.; Vachet, R.W. Protein Surface Mapping Using Diethylpyrocarbonate with Mass Spectrometric Detection. Anal. Chem. 2008, 80, 2895–2904. [Google Scholar] [CrossRef] [Green Version]
- Narindrasorasak, S.; Kulkarni, P.; Deschamps, P.; She, Y.-M.; Sarkar, B. Characterization and Copper Binding Properties of Human COMMD1 (MURR1). Biochemistry 2007, 46, 3116–3128. [Google Scholar] [CrossRef]
- Qin, K.; Yang, Y.; Mastrangelo, P.; Westaway, D. Mapping Cu(II) Binding Sites in Prion Proteins by Diethyl Pyrocarbonate Modification and Matrix-assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) Mass Spectrometric Footprinting. J. Biol. Chem. 2002, 277, 1981–1990. [Google Scholar] [CrossRef] [Green Version]
- Walter, E.D.; Stevens, D.J.; Visconte, M.P.; Millhauser, G.L. The Prion Protein is a Combined Zinc and Copper Binding Protein: Zn2+ Alters the Distribution of Cu2+ Coordination Modes. J. Am. Chem. Soc. 2007, 129, 15440–15441. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S.; Das, K.P. Identification of Histidine Residues Involved in Zn2+ Binding to αA- and αB-Crystallin by Chemical Modification and MALDI TOF Mass Spectrometry. Protein J. 2012, 31, 623–640. [Google Scholar] [CrossRef]
- Piotrowski, C.; Moretti, R.; Ihling, C.H.; Haedicke, A.; Liepold, T.; Lipstein, N.; Meiler, J.; Jahn, O.; Sinz, A. Delineating the Molecular Basis of the Calmodulin–bMunc13-2 Interaction by Cross-Linking/Mass Spectrometry—Evidence for a Novel CaM Binding Motif in bMunc13-2. Cells 2020, 9, 136. [Google Scholar] [CrossRef] [Green Version]
- Lipstein, N.; Schaks, S.; Dimova, K.; Kalkhof, S.; Ihling, C.; Kolbel, K.; Ashery, U.; Rhee, J.; Brose, N.; Sinz, A.; et al. Nonconserved Ca2+/Calmodulin Binding Sites in Munc13s Differentially Control Synaptic Short-Term Plasticity. Mol. Cell. Biol. 2012, 32, 4628–4641. [Google Scholar] [CrossRef] [Green Version]
- Dimova, K.; Kawabe, H.; Betz, A.; Brose, N.; Jahn, O. Characterization of the Munc13-calmodulin interaction by photoaffinity labeling. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, C.; Götze, M.; Piotrowski, C.; Arlt, C.; Rehkamp, A.; Ihling, C.; Hage, C.; Sinz, A. Carboxyl-Photo-Reactive MS-Cleavable Cross-Linkers: Unveiling a Hidden Aspect of Diazirine-Based Reagents. Anal. Chem. 2018, 90, 2805–2809. [Google Scholar] [CrossRef]
- Piersimoni, L.; Kastritis, P.L.; Arlt, C.; Sinz, A. Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein–Protein Interactions—A Method for All Seasons. Chem. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wen, J.; Huang, R.Y.-C.; Blankenship, R.E.; Gross, M.L. Mass spectrometry-based carboxyl footprinting of proteins: Method evaluation. Int. J. Mass Spectrom. 2012, 312, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gau, B.C.; Jones, L.M.; Vidavsky, I.; Gross, M.L. Fast Photochemical Oxidation of Proteins for Comparing Structures of Protein−Ligand Complexes: The Calmodulin−Peptide Model System. Anal. Chem. 2011, 83, 311–318. [Google Scholar] [CrossRef]
- Ikura, M.; Clore, G.M.; Gronenborn, A.M.; Zhu, G.; Klee, C.B.; Bax, A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 1992, 256, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Wan, K.X.; Vidavsky, I.; Gross, M.L. Comparing similar spectra: From similarity index to spectral contrast angle. J. Am. Soc. Mass Spectrom. 2002, 13, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.M.; Rempel, D.L.; Zhao, J.; Giblin, D.E.; Gross, M.L. Probing Ca2+-Induced Conformational Changes in Porcine Calmodulin by H/D Exchange and ESI-MS: Effect of Cations and Ionic Strength. Biochemistry 2003, 42, 15388–15397. [Google Scholar] [CrossRef] [PubMed]
- Linse, S.; Helmersson, A.; Forsén, S. Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 1991, 266, 8050–8054. [Google Scholar] [CrossRef]
- Sperry, J.B.; Huang, R.Y.-C.; Zhu, M.M.; Rempel, D.L.; Gross, M.L. Hydrophobic peptides affect binding of calmodulin and Ca2+ as explored by H/D amide exchange and mass spectrometry. Int. J. Mass Spectrom. 2011, 302, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.R.; Zhang, M.M.; Rempel, D.L.; Gross, M.L. A Single Approach Reveals the Composite Conformational Changes, Order of Binding, and Affinities for Calcium Binding to Calmodulin. Anal. Chem. 2019, 91, 5508–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.R.; Rempel, D.L.; Gross, M.L. Composite Conformational Changes of Signaling Proteins upon Ligand Binding Revealed by a Single Approach: Calcium-Calmodulin Study. Anal. Chem. 2019, 91, 12560–12567. [Google Scholar] [CrossRef] [PubMed]
- Zygiel, E.M.; Nolan, E.M. Transition Metal Sequestration by the Host-Defense Protein Calprotectin. Annu. Rev. Biochem. 2018, 87, 621–643. [Google Scholar] [CrossRef] [PubMed]
- Brophy, M.B.; Hayden, J.A.; Nolan, E.M. Calcium Ion Gradients Modulate the Zinc Affinity and Antibacterial Activity of Human Calprotectin. J. Am. Chem. Soc. 2012, 134, 18089–18100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korndörfer, I.P.; Brueckner, F.; Skerra, A. The Crystal Structure of the Human (S100A8/S100A9)2 Heterotetramer, Calprotectin, Illustrates how Conformational Changes of Interacting α-Helices Can Determine Specific Association of Two EF-hand Proteins. J. Mol. Biol. 2007, 370, 887–898. [Google Scholar] [CrossRef]
- Strupat, K.; Rogniaux, H.; Van Dorsselaer, A.; Roth, J.; Vogl, T. Calcium-induced noncovalently linked tetramers of MRP8 and MRP14 are confirmed by electrospray ionization-mass analysis. J. Am. Soc. Mass Spectrom. 2000, 11, 780–788. [Google Scholar] [CrossRef]
- Gagnon, D.M.; Brophy, M.B.; Bowman, S.E.J.; Stich, T.A.; Drennan, C.L.; Britt, R.D.; Nolan, E.M. Manganese Binding Properties of Human Calprotectin under Conditions of High and Low Calcium: X-ray Crystallographic and Advanced Electron Paramagnetic Resonance Spectroscopic Analysis. J. Am. Chem. Soc. 2015, 137, 3004–3016. [Google Scholar] [CrossRef]
- Nakashige, T.G.; Zygiel, E.M.; Drennan, C.L.; Nolan, E.M. Nickel Sequestration by the Host-Defense Protein Human Calprotectin. J. Am. Chem. Soc. 2017, 139, 8828–8836. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Ottolini, D.; Calì, T.; Carafoli, E. Calcium in Health and Disease. In Metal Ions in Life Sciences; Springer: Dordrecht, The Netherland, 2013; pp. 81–137. [Google Scholar]
- Leukert, N.; Vogl, T.; Strupat, K.; Reichelt, R.; Sorg, C.; Roth, J. Calcium-dependent Tetramer Formation of S100A8 and S100A9 is Essential for Biological Activity. J. Mol. Biol. 2006, 359, 961–972. [Google Scholar] [CrossRef]
- Gomes, F.P.; Yates, J.R. Recent trends of capillary electrophoresis-mass spectrometry in proteomics research. Mass Spectrom. Rev. 2019, 38, 445–460. [Google Scholar] [CrossRef]
- Edelmann, M.J. Strong Cation Exchange Chromatography in Analysis of Posttranslational Modifications: Innovations and Perspectives. J. Biomed. Biotechnol. 2011, 2011, 936508. [Google Scholar] [CrossRef]
- Cavanagh, J.; Benson, L.M.; Thompson, R.; Naylor, S. In-Line Desalting Mass Spectrometry for the Study of Noncovalent Biological Complexes. Anal. Chem. 2003, 75, 3281–3286. [Google Scholar] [CrossRef]
- Farsang, E.; Guillarme, D.; Veuthey, J.-L.; Beck, A.; Lauber, M.; Schmudlach, A.; Fekete, S. Coupling non-denaturing chromatography to mass spectrometry for the characterization of monoclonal antibodies and related products. J. Pharm. Biomed. Anal. 2020, 185, 113207. [Google Scholar] [CrossRef] [PubMed]
- Pascal, B.D.; Willis, S.; Lauer, J.L.; Landgraf, R.R.; West, G.M.; Marciano, D.; Novick, S.; Goswami, D.; Chalmers, M.J.; Griffin, P.R. HDX Workbench: Software for the Analysis of H/D Exchange MS Data. J. Am. Soc. Mass Spectrom. 2012, 23, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.M.C.; Ahdash, Z.; Martens, C.; Politis, A. Deuteros: Software for rapid analysis and visualization of data from differential hydrogen deuterium exchange-mass spectrometry. Bioinformatics 2019, 35, 3171–3173. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, Y.; Coales, S.J.; Southern, M.R.; Nemeth-Cawley, J.F.; Stranz, D.D.; Griffin, P.R. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J. Biomol. Tech. 2003, 14, 171–182. [Google Scholar] [PubMed]
- Hageman, T.S.; Weis, D.D. Reliable Identification of Significant Differences in Differential Hydrogen Exchange-Mass Spectrometry Measurements Using a Hybrid Significance Testing Approach. Anal. Chem. 2019, 91, 8008–8016. [Google Scholar] [CrossRef]
- Hageman, T.S.; Weis, D.D. A Structural Variant Approach for Establishing a Detection Limit in Differential Hydrogen Exchange-Mass Spectrometry Measurements. Anal. Chem. 2019, 91, 8017–8024. [Google Scholar] [CrossRef]
- Hageman, T.S.; Wrigley, M.S.; Weis, D.D. Statistical Equivalence Testing of Higher-Order Protein Structures with Differential Hydrogen Exchange-Mass Spectrometry (HX-MS). Anal. Chem. 2021, 93, 6980–6988. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Gross, M.L. Mass Spectrometry-Based Structural Proteomics for Metal Ion/Protein Binding Studies. Biomolecules 2022, 12, 135. https://doi.org/10.3390/biom12010135
Lin Y, Gross ML. Mass Spectrometry-Based Structural Proteomics for Metal Ion/Protein Binding Studies. Biomolecules. 2022; 12(1):135. https://doi.org/10.3390/biom12010135
Chicago/Turabian StyleLin, Yanchun, and Michael L. Gross. 2022. "Mass Spectrometry-Based Structural Proteomics for Metal Ion/Protein Binding Studies" Biomolecules 12, no. 1: 135. https://doi.org/10.3390/biom12010135
APA StyleLin, Y., & Gross, M. L. (2022). Mass Spectrometry-Based Structural Proteomics for Metal Ion/Protein Binding Studies. Biomolecules, 12(1), 135. https://doi.org/10.3390/biom12010135